CRISPR vs. TALENs vs. ZFNs: A 2025 Specificity and Application Comparison for Researchers
This article provides a comprehensive, up-to-date comparison of the specificity of the three primary genome-editing platforms: CRISPR, TALENs, and ZFNs.
CRISPR vs. TALENs vs. ZFNs: A 2025 Specificity and Application Comparison for Researchers
Abstract
This article provides a comprehensive, up-to-date comparison of the specificity of the three primary genome-editing platforms: CRISPR, TALENs, and ZFNs. Tailored for researchers, scientists, and drug development professionals, it explores the fundamental mechanisms that dictate precision, delves into current therapeutic and research applications, and addresses key challenges like off-target effects. The content synthesizes the latest 2025 clinical data and technological advancements, including base editing and AI-driven design, to offer a clear, evidence-based framework for selecting the optimal tool for specific research and clinical goals.
Understanding the Core Mechanisms: How CRISPR, TALENs, and ZFNs Achieve Specificity
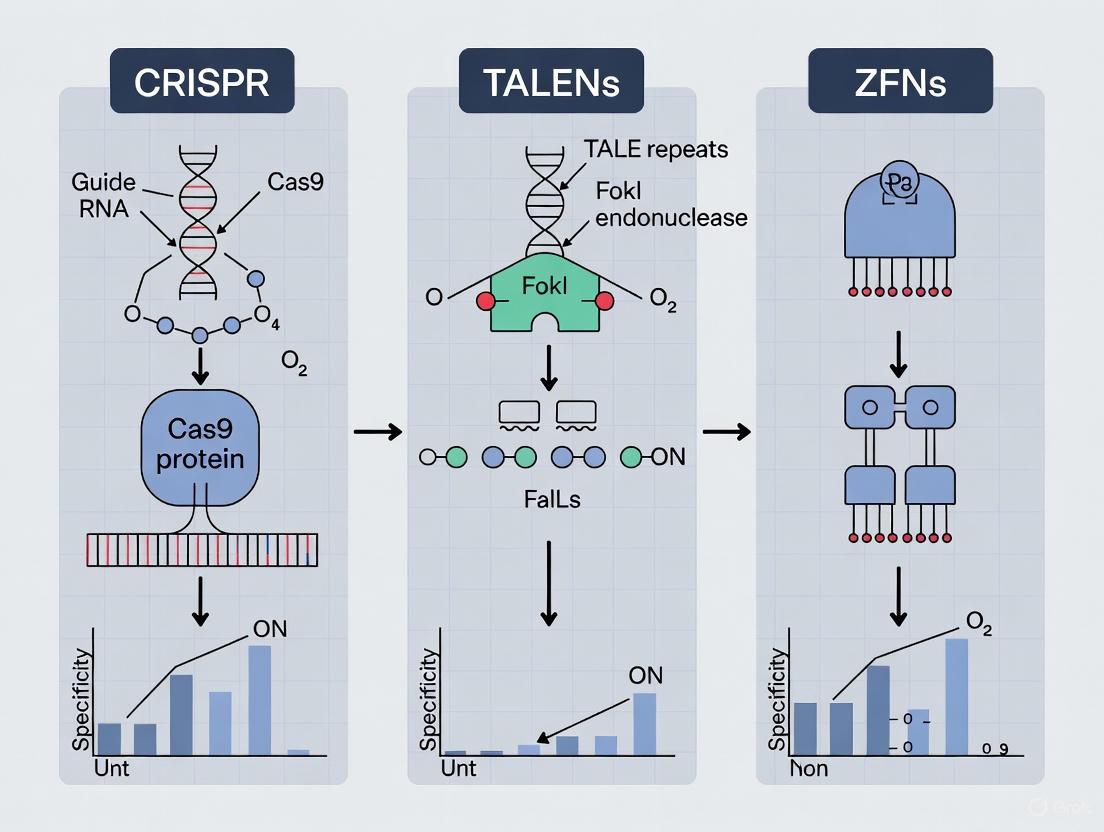
The field of genome engineering has been revolutionized by technologies that enable precise modifications to DNA sequences. These tools can be fundamentally categorized by their targeting mechanisms: those that rely on protein-DNA interactions and those that utilize RNA-guided DNA recognition [1] [2]. Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs) belong to the first category, where engineered proteins directly bind to specific DNA sequences. In contrast, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) system represents the latter, using a guide RNA molecule to direct a nuclease to its DNA target [3] [2]. This fundamental distinction in targeting philosophy has profound implications for the design, specificity, efficiency, and application of these technologies in research and therapy. This guide provides an objective comparison of these systems, focusing on their performance and the experimental data that defines their capabilities.
Protein-DNA Interaction Systems
Zinc Finger Nucleases (ZFNs) were among the first programmable genome editing tools. Their design involves fusing a engineered zinc finger DNA-binding domain to the FokI nuclease domain [1] [4]. Each zinc finger module typically recognizes a 3-base pair sequence, and arrays of multiple fingers are assembled to create a protein that binds a specific 9-18 bp sequence [1]. Because the FokI nuclease must dimerize to become active, a pair of ZFNs is required to bind opposite strands of DNA, with their cleavage sites facing each other across a short spacer region [1].
Transcription Activator-Like Effector Nucleases (TALENs) operate on a similar principle but use a different DNA-binding architecture derived from TALE proteins in Xanthomonas bacteria [1] [5]. The DNA-binding domain of a TALEN consists of a series of 33-35 amino acid repeats, each recognizing a single base pair through two hypervariable amino acids known as Repeat Variable Diresidues (RVDs) [1] [5]. Common RVDs include NI for adenine, HD for cytosine, NN for guanine/adenine, and NG for thymine [5]. Like ZFNs, TALENs also use the FokI nuclease domain and require pairing for activity.
RNA-Guided Systems
The CRISPR-Cas system represents a paradigm shift from protein-based targeting. This system originates from a prokaryotic adaptive immune mechanism that protects bacteria from viral infections [2] [6]. The most widely used variant, CRISPR-Cas9, consists of two key components: the Cas9 nuclease and a guide RNA (gRNA) [2]. The gRNA is a synthetic fusion of two natural RNAs—crRNA and tracrRNA—and contains a ~20 nucleotide sequence that is complementary to the target DNA site [2]. Cas9 is directed to the target DNA by RNA-DNA base pairing, and requires a short Protospacer Adjacent Motif (PAM) sequence adjacent to the target site for recognition [2]. For Cas9 from Streptococcus pyogenes, the PAM is 5'-NGG-3' [2].
The diagram below illustrates the fundamental differences in how these three systems locate and bind their DNA targets.
Comparative Performance Analysis
Specificity and Off-Target Effects
Specificity—the ability to edit only the intended target—is a critical parameter for evaluating genome editing tools. Each technology has distinct strengths and limitations in this regard.
TALENs are renowned for their high specificity, which stems from their long, highly specific DNA-binding domains (typically 30-40 bp per TALEN pair) and the requirement for dimerization of FokI nuclease [3]. Research has shown that TALEN specificity can be further enhanced by using non-conventional RVDs (ncRVDs) that improve discrimination between similar DNA sequences [5]. For instance, in one study, researchers engineered TALENs with ncRVDs to distinguish between the highly similar HBB and HBD genes (94% identity), successfully reducing off-target cleavage while maintaining on-target activity [5].
ZFNs also exhibit high specificity due to their relatively long recognition sites (18-36 bp for a ZFN pair) and the FokI dimerization requirement [1] [3]. However, ZFN design can be complicated by context-dependent effects between adjacent zinc fingers, which can affect DNA-binding specificity [1]. The potential for off-target effects exists if ZFNs bind and cleave at sites with similar sequences, though proper design minimizes this risk.
CRISPR-Cas9 systems have raised more concerns about off-target effects because the gRNA can tolerate mismatches, particularly in the 5' region distal to the PAM sequence [3] [2]. However, the off-target effects of CRISPR are generally considered more predictable than those of ZFNs and TALENs because they primarily depend on sequence homology [2]. Numerous strategies have been developed to improve CRISPR specificity, including the use of high-fidelity Cas9 variants, truncated gRNAs with enhanced specificity, and dual nickase systems that require two adjacent gRNAs for double-strand break formation [2] [6].
Quantitative Comparison of Editing Efficiency and Practical Parameters
The table below summarizes key performance metrics and practical characteristics of the three genome editing technologies, based on comparative experimental data.
Table 1: Performance Comparison of Major Genome Editing Technologies
| Parameter | ZFN | TALEN | CRISPR-Cas |
|---|---|---|---|
| Targeting Mechanism | Protein-DNA interaction [1] | Protein-DNA interaction [1] | RNA-DNA hybridization [2] |
| Typical Target Size | 18-36 bp per ZFN pair [2] | 30-40 bp per TALEN pair [2] | 22 bp [2] |
| Editing Efficiency | 0%-12% (low) [2] | 0%-76% (moderate) [2] | 0%-81% (high) [2] |
| Ease of Design | Difficult, requires protein engineering for each target [3] [2] | Difficult, requires protein engineering for each target [3] [2] | Easy, only requires changing gRNA sequence [3] [2] |
| Multiplexing Potential | Less feasible [2] | Less feasible [2] | Highly feasible [2] |
| Off-Target Prediction | Less predictable [2] | Less predictable [2] | Highly predictable [2] |
| Construction Cost & Time | High cost, time-consuming [3] | High cost, time-consuming [3] | Low cost, rapid [3] |
| Common Delivery Methods | AAV, viral vectors [4] [2] | AAV, viral vectors [2] | AAV, lentivirus, non-viral methods [7] [2] |
Experimental Approaches for Assessing Specificity
Methodologies for Evaluating TALEN Specificity
The high specificity of TALENs has been systematically validated through rigorous experimental approaches. One comprehensive study employed a library-based high-throughput screen of TALENs containing non-conventional RVDs (ncRVDs) [5]. The experimental workflow involved:
- Library Construction: Creating a library of degenerate RVDs randomized at positions 12 and 13 using short overlap PCR with NNK codon degeneracy, then incorporating these RVDs into TALEN arrays via solid-phase assembly [5].
- Specificity Profiling: Screening approximately 18,000 TALEN-target combinations in a yeast-based assay system to characterize the cleavage activity and specificity of each ncRVD variant [5].
- Hierarchical Clustering: Analyzing cleavage profiles to identify clusters of RVDs with similar specificity patterns, revealing that the identity of amino acids at positions 12 and 13 has a more pronounced impact on nuclease activity and specificity than the RVD's position in the array [5].
- Validation in Mammalian Cells: Testing selected TALENs with optimized ncRVDs in mammalian cells to demonstrate their ability to discriminate between highly similar target sequences, such as the HBB and HBD genes (94% identity) [5].
This systematic approach identified ncRVDs with enhanced discriminatory power, enabling the design of TALENs that minimize off-target effects while maintaining robust on-target activity—a crucial consideration for therapeutic applications [5].
CRISPR-Cas9 Specificity Assessment
Multiple methods have been developed to evaluate and improve CRISPR-Cas9 specificity:
- In Silico Prediction: Computational tools predict potential off-target sites based on sequence similarity to the intended target, particularly in the seed region near the PAM sequence [2].
- Genome-Wide Assays: Techniques such as CIRCLE-seq, GUIDE-seq, and BLESS directly capture off-target cleavage events across the entire genome, providing experimental validation of specificity [2].
- High-Fidelity Variants: Engineered Cas9 proteins with reduced off-target activity, such as eSpCas9 and SpCas9-HF1, have been developed through rational design to minimize non-specific interactions with DNA [2] [6].
- Dual Nickase Systems: Using two Cas9 nickases with paired gRNAs to create staggered cuts rather than double-strand breaks significantly enhances specificity, as off-target sites are unlikely to be cleaved by both nickases simultaneously [2].
Research Reagent Solutions
The following table outlines essential reagents and tools used in genome editing research, providing researchers with a practical resource for experimental planning.
Table 2: Essential Research Reagents for Genome Editing Studies
| Reagent/Tool | Function | Technology |
|---|---|---|
| CompoZr ZFN Platform | Pre-designed, validated ZFNs for specific genomic targets [1] | ZFN |
| TALE Repeat Kits | Molecular cloning kits for assembling custom TALE arrays (e.g., Golden Gate assembly) [1] | TALEN |
| Cas9 Expression Vectors | Plasmids encoding Cas9 nuclease with various promoters for different cell types [2] | CRISPR |
| Guide RNA Cloning Systems | Backbone vectors for rapid insertion of target-specific guide RNA sequences [2] | CRISPR |
| High-Fidelity Cas9 Variants | Engineered Cas9 proteins with reduced off-target effects (e.g., eSpCas9, SpCas9-HF1) [2] [6] | CRISPR |
| AAV Delivery Vectors | Adeno-associated virus serotypes optimized for efficient delivery of editing components [7] [4] | All |
| Off-Target Assessment Kits | Commercial kits for genome-wide identification of off-target sites (e.g., GUIDE-seq) [2] | All |
The fundamental distinction between RNA-guided and protein-DNA interaction mechanisms underlies the practical differences in specificity, efficiency, and ease-of-use among major genome editing technologies. TALENs offer high specificity due to their long recognition sequences and tunable RVDs, making them suitable for applications where precision is paramount. ZFNs, while historically important, present greater design challenges. CRISPR-Cas systems provide unprecedented flexibility and ease of design, with continuously improving specificity through engineered variants and optimized protocols.
The choice between these systems depends on the specific research requirements: TALENs for high-precision applications with minimal off-target effects, and CRISPR for multiplexed experiments and projects requiring rapid implementation. As these technologies continue to evolve, particularly with the emergence of base editing and prime editing systems, the landscape of precision genome editing will continue to offer increasingly sophisticated solutions for research and therapeutic development.
The advent of programmable nucleases has revolutionized genetic engineering, enabling precise modifications to DNA sequences within living cells. Among the most significant technologies in this domain are Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the CRISPR-Cas9 system. Each platform employs a distinct structural approach to achieve DNA recognition and cleavage. ZFNs and TALENs rely on engineered protein domains to directly interact with DNA sequences, while CRISPR-Cas9 utilizes a guide RNA (gRNA) molecule to direct a nuclease enzyme to a specific genomic locus. Understanding the molecular architecture of these systems—specifically the composition of zinc finger arrays, TALE repeats, and gRNA—is critical for selecting the appropriate tool for specific research or therapeutic applications. These architectures directly influence key performance parameters including specificity, efficiency, ease of design, and potential for off-target effects [8] [9]. This guide provides a structural comparison of these three systems, supported by experimental data and detailed methodologies, to inform researchers and drug development professionals.
Molecular Architecture and Design
Zinc Finger Arrays (ZFNs)
Zinc Finger Nucleases (ZFNs) are synthetic proteins created by fusing a custom-designed zinc finger array to the catalytic domain of the FokI restriction enzyme. The DNA-binding domain consists of multiple zinc finger motifs, each comprising approximately 30 amino acids folded around a zinc ion. A fundamental aspect of their architecture is that each individual zinc finger motif recognizes and binds to a specific 3-base pair (bp) DNA triplet [10] [9]. To target a unique genomic sequence, researchers engineer an array of multiple fingers (typically 3 to 6) in tandem, creating a protein domain that recognizes a contiguous 9 to 18 bp sequence. Since FokI requires dimerization to become active, a pair of ZFNs must be designed to bind opposite strands of the DNA, flanking the intended cleavage site. The cleavage event occurs in the spacer region between the two binding sites [9].
The primary challenge with ZFN technology lies in its complex design process. Achieving high-affinity and specific binding often requires extensive optimization, as the DNA-binding specificity of individual zinc fingers can be influenced by context—neighboring fingers can affect each other's binding preferences. This context-dependent specificity makes the rational design of ZFNs challenging and time-consuming, often requiring specialized expertise and sophisticated screening methods to develop effective nucleases [10] [11] [9].
TALE Repeats (TALENs)
Transcription Activator-Like Effector Nucleases (TALENs) are also fusion proteins, combining a DNA-binding domain derived from TAL effectors (proteins from plant pathogens) with the FokI nuclease domain. The DNA-binding domain is composed of highly conserved TALE repeats, each typically 33-35 amino acids in length [9]. A key breakthrough that made TALENs a transformative technology was the discovery of a simple, modular code for DNA recognition: within each repeat, two variable amino acids at positions 12 and 13, known as the Repeat-Variable Diresidue (RVD), determine nucleotide specificity [10] [9].
Common RVDs and their recognized nucleotides include:
- NI for Adenine (A)
- NG for Thymine (T)
- HD for Cytosine (C)
- NN for Guanine (G) or Adenine (A) [10] [9]
This one-to-one correspondence between RVDs and DNA bases makes TALEN design highly predictable and straightforward compared to ZFNs. Researchers can essentially string together a series of TALE repeats in an order that mirrors the target DNA sequence. Like ZFNs, TALENs function as pairs, with two TALEN proteins binding to opposite DNA strands and the FokI domains dimerizing to create a double-strand break in the intervening spacer region. While the design is more modular, a practical limitation is the large size of the TALEN coding sequence and the repetitive nature of the TALE repeats, which can complicate viral packaging and introduce challenges in cloning [10] [11].
Guide RNA (gRNA) in CRISPR-Cas9
The CRISPR-Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9) system functions through a fundamentally different mechanism. Its specificity is conferred not by a protein domain, but by a short guide RNA (gRNA) [8]. The gRNA is a synthetic chimera composed of a CRISPR RNA (crRNA) derivative, which contains a ~20 nucleotide sequence complementary to the target DNA, and a trans-activating crRNA (tracrRNA), which serves as a scaffold for binding the Cas9 nuclease [10]. The gRNA directs the Cas9 protein to the target site through Watson-Crick base pairing between its spacer sequence and the genomic DNA.
A critical requirement for Cas9 recognition and cleavage is the presence of a short Protospacer Adjacent Motif (PAM) sequence immediately downstream of the target site. The most commonly used Cas9 from Streptococcus pyogenes (SpCas9) requires a 5'-NGG-3' PAM sequence, where "N" can be any nucleotide [10]. Upon binding to a target sequence that is complementary to the gRNA and adjacent to a valid PAM, the Cas9 protein undergoes a conformational change that activates its two nuclease domains (HNH and RuvC), generating a blunt-ended double-strand break [8] [10].
The following diagram illustrates the fundamental architectural differences and the common double-strand break repair pathways shared by these technologies.
Performance Comparison and Experimental Data
Direct comparative studies provide the most valuable insights for tool selection. The following table summarizes key performance metrics for ZFNs, TALENs, and CRISPR-Cas9, drawing from multiple sources, including a seminal parallel comparison using the GUIDE-seq method for detecting off-target effects [12] [13] [8].
| Feature | CRISPR-Cas9 (SpCas9) | TALENs | ZFNs |
|---|---|---|---|
| Targeting Molecule | Guide RNA (gRNA) | TALE Protein Repeats | Zinc Finger Protein Array |
| Recognition Code | RNA-DNA base pairing (~20 nt) | 1 RVD : 1 DNA base | 1 finger : ~3 DNA bases |
| Specificity (Off-Targets) | Variable; can be high with optimized systems [13] | Generally high [13] [11] | Can be significant; design-dependent [13] |
| Efficiency (Knockout) | High [12] [8] | Moderate to High [8] [11] | Moderate [8] |
| Ease of Design & Use | Very high (designing a gRNA is simple) [8] [10] | Moderate (requires protein engineering) [8] [10] | Low (complex, context-dependent design) [8] [10] |
| Time to Develop Reagents | Days [8] | Days to weeks [10] | Months [10] |
| Multiplexing Capacity | High (multiple gRNAs easily designed) [8] | Low (difficult to engineer multiple proteins) [8] | Low (difficult to engineer multiple proteins) [8] |
| Typical Cost | Low [8] [10] | High [8] [10] | Very High [8] [10] |
| Key Limitation | PAM requirement, off-target effects [8] [10] | Large size, repetitive sequence, delivery challenges [10] [11] | Difficult, time-consuming design; potential cytotoxicity [10] [11] |
A critical 2021 study provided a direct, parallel comparison of the three nuclease platforms using the GUIDE-seq method to genome-widely profile off-target effects in the context of human papillomavirus (HPV) gene therapy [13]. The results were striking:
- In the HPV URR region, SpCas9 had 0 off-target sites detected, compared to 1 for TALENs and 287 for one ZFN pair [13].
- In the E6 region, SpCas9 had 0 off-targets, while TALENs had 7 [13].
- In the E7 region, SpCas9 had 4 off-targets, which was still significantly fewer than the 36 detected for TALENs [13].
This study concluded that for their specific application, SpCas9 was both more efficient and specific than ZFNs and TALENs [13]. It is important to note that CRISPR specificity continues to improve with the development of high-fidelity Cas9 variants.
Beyond specificity, survey data from the drug discovery sector reveals practical workflow differences. Generating a CRISPR knockout cell line takes a median of 3 months, while a knock-in takes 6 months. Furthermore, researchers often must repeat the entire CRISPR workflow a median of 3 times before achieving success, highlighting that even the most user-friendly system involves a tedious and time-consuming process [12].
Detailed Experimental Protocols
To ensure the reproducibility of comparative studies and the validation of nuclease performance, standardized experimental protocols are essential. Below are detailed methodologies for two key assays used in the cited research.
GUIDE-seq for Genome-Wide Off-Target Detection
The GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) method is a powerful and sensitive technique for detecting off-target nuclease activity without prior knowledge of potential sites [13].
Principle: A short, double-stranded oligonucleotide tag is incorporated into double-strand breaks (DSBs) generated by the nuclease in living cells. These tagged sites are then enriched and identified via next-generation sequencing.
Key Reagents:
- Oligonucleotide Tag: A short, blunt-ended, double-stranded DNA oligo with phosphorothioate linkages on the ends to resist exonuclease digestion.
- Transfection Reagent: For delivering the nuclease constructs and the oligonucleotide tag into the target cells.
- PCR Primers: Specific to the oligonucleotide tag for amplification of integrated sites.
- Next-Generation Sequencing Platform.
Procedure:
- Co-transfection: Co-deliver the nuclease expression plasmid (or ribonucleoprotein complex for CRISPR) and the GUIDE-seq oligonucleotide tag into the target cells.
- Incubation: Allow cells to grow for 48-72 hours to permit nuclease activity and tag integration.
- Genomic DNA Extraction: Harvest cells and extract high-molecular-weight genomic DNA.
- Tag-Specific PCR: Shear the genomic DNA and perform PCR using a biotinylated primer specific to the integrated oligo tag.
- Library Preparation and Sequencing: Purify the PCR amplicons, prepare a sequencing library, and sequence on an NGS platform.
- Bioinformatic Analysis: Map the sequencing reads back to the reference genome to identify all genomic locations where the tag was integrated, which correspond to nuclease-induced DSBs. Compare these sites to the intended on-target sequence to catalog off-target events [13].
Cell Viability and Editing Efficiency Assay
This protocol assesses the functional efficiency and cytotoxicity of nucleases, which is crucial for therapeutic applications.
Principle: The rate of targeted mutagenesis is measured at the on-target site, while cell survival and health are monitored to evaluate the toxicity associated with nuclease expression.
Key Reagents:
- Nuclease Expression Vectors: Plasmids or mRNA encoding ZFNs, TALENs, or CRISPR-Cas9/gRNA.
- Control Vectors: Inactive nuclease controls (e.g., catalytically dead Cas9 for CRISPR).
- Cell Culture Reagents.
- Genomic DNA Extraction Kit.
- T7 Endonuclease I or Surveyor Nuclease: For detecting mismatches in heteroduplex DNA caused by NHEJ repair.
- PCR Reagents and Primers flanking the target site.
- Cell Viability Assay Kit (e.g., MTT, CellTiter-Glo).
Procedure:
- Cell Transfection: Introduce nuclease constructs into the chosen cell model (e.g., immortalized cell lines, primary cells). Include controls transfected with inactive nucleases and non-transfected cells.
- Viability Measurement: At 72-96 hours post-transfection, assay cell viability using a method like ATP quantification (CellTiter-Glo). Normalize viability to control treatments to determine nuclease-associated toxicity [12] [11].
- Harvest Genomic DNA: Extract genomic DNA from a portion of the transfected cell population.
- Amplify Target Locus: Perform PCR to amplify the genomic region surrounding the nuclease target site.
- Detect Indel Mutations:
- T7E1/Surveyor Assay: Denature and reanneal the PCR products to form heteroduplexes. Digestion with the mismatch-specific nuclease will cleave the DNA if indels are present. Analyze the cleavage products by gel electrophoresis. The ratio of cleaved to uncleaved products allows for estimation of editing efficiency [9].
- Data Analysis: Calculate the percentage of indels and correlate this with cell viability data to determine the therapeutic index of the nuclease.
The Scientist's Toolkit: Essential Research Reagents
Successful gene editing experiments require a suite of specialized reagents and tools. The following table details key solutions and their functions for working with these nuclease platforms.
| Research Reagent / Solution | Function & Application |
|---|---|
| Guide RNA (gRNA) Expression Plasmid | A vector for expressing the custom ~20 nt gRNA within cells; the core of CRISPR experiment design. |
| Cas9 Expression Plasmid or mRNA | Delivers the Cas9 nuclease; can be co-delivered with gRNA plasmid or used as a pre-complexed Ribonucleoprotein (RNP). |
| TALEN Expression Kit | Modular kits (e.g., Golden Gate assembly) containing TALE repeat modules for efficient construction of custom TALEN pairs. |
| ZFN Expression Vector | Plasmids for expressing the pairs of engineered zinc finger proteins fused to FokI nuclease. |
| GUIDE-seq Oligo Duplex | The double-stranded oligonucleotide tag used for genome-wide, unbiased identification of off-target cleavage sites. |
| T7 Endonuclease I / Surveyor Nuclease | Enzymes used for the rapid detection and quantification of indel mutations at the target site. |
| HDR Donor Template | Single-stranded oligonucleotide (ssODN) or double-stranded DNA vector containing the desired edit flanked by homology arms, used for precise knock-in via HDR. |
| Cell Line-Specific Transfection Reagent | Chemical or lipid-based reagents for efficient delivery of nuclease constructs (DNA, RNA, or RNP) into target cells. |
| NGS-Based Off-Target Analysis Service | Commercial services that use methods like GUIDE-seq or CIRCLE-seq to provide a comprehensive report of nuclease off-target activity. |
The structural deep dive into gRNA, TALE repeats, and zinc finger arrays reveals a clear trade-off between simplicity and precision. CRISPR-Cas9, with its RNA-guided architecture, offers an unparalleled combination of ease of design, high efficiency, and multiplexing capability, making it the dominant tool for most high-throughput and research applications [12] [8]. However, its specificity is contingent on gRNA design and can be prone to off-target effects, though high-fidelity variants are mitigating this issue [13] [8].
TALENs, with their modular protein-DNA code, provide high specificity and are effective in targeting complex regions, making them valuable for therapeutic applications where minimizing off-targets is paramount [13] [11]. Their main drawbacks are the larger size and more labor-intensive protein engineering process.
ZFNs, as the pioneering technology, demonstrated the feasibility of programmable gene editing but are now largely superseded due to their complex, context-dependent design process and higher potential for cytotoxicity and off-target effects [13] [10] [11].
The choice among these platforms is not one-size-fits-all. For rapid screening and multiplexed experiments, CRISPR is often the optimal choice. For clinical applications requiring the highest possible specificity and where delivery constraints can be overcome, TALENs remain a powerful alternative. The continued advancement of all three platforms, including the development of base editors and prime editors, ensures that the gene editing toolkit will continue to expand, offering researchers and clinicians increasingly sophisticated instruments for precise genetic manipulation.
The Critical Role of PAM Sequences in CRISPR and Their Impact on Targetable Sites
The advent of programmable genome editing technologies has revolutionized biological research and therapeutic development. Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the CRISPR-Cas9 system represent three generations of genome editing tools that enable precise modifications to DNA sequences [14]. These technologies function by creating targeted double-strand breaks (DSBs) in the DNA, which are subsequently repaired by the cell's endogenous repair mechanisms—either error-prone non-homologous end joining (NHEJ) or homology-directed repair (HDR) [15] [16]. While all three systems achieve the same fundamental goal of targeted genome modification, they differ dramatically in their molecular architectures, recognition mechanisms, and practical implementation. The CRISPR-Cas9 system, in particular, has gained widespread adoption due to its simplicity and versatility, but its targeting capabilities are constrained by a unique requirement: the protospacer adjacent motif (PAM) sequence [17] [18]. This PAM requirement represents a critical differentiator between CRISPR and earlier technologies, with profound implications for targetable sites in the genome, experimental design, and therapeutic applications.
The comparative analysis of these editing technologies extends beyond mere mechanism to encompass critical performance metrics including specificity, efficiency, and ease of design. TALENs employ a pair of customizable DNA-binding proteins derived from plant pathogens fused to the FokI nuclease domain, with each DNA-binding module recognizing a single nucleotide [19] [16]. ZFNs similarly utilize FokI nuclease but rely on zinc finger protein arrays that typically recognize triplets of nucleotides [19] [14]. In contrast, the CRISPR-Cas9 system depends on a complex between the Cas9 nuclease and a short guide RNA (sgRNA) that hybridizes with the target DNA through Watson-Crick base pairing [17] [14]. This fundamental difference in recognition mechanism—protein-DNA for ZFNs and TALENs versus RNA-DNA for CRISPR—underlies their distinct strengths and limitations, particularly regarding PAM constraints and targeting flexibility.
The Protospacer Adjacent Motif (PAM): A Critical CRISPR-Specific Requirement
Biological Function and Definition
The protospacer adjacent motif (PAM) is a short, specific DNA sequence (typically 2-6 base pairs in length) that immediately follows the DNA region targeted for cleavage by the CRISPR-Cas system [17] [18]. This motif serves as an essential recognition element that enables Cas nucleases to distinguish between self and non-self DNA, thereby preventing autoimmunity in bacterial CRISPR systems [17] [20]. In nature, when bacteria survive viral infection, they incorporate fragments of viral DNA (protospacers) into their CRISPR arrays as spacers [17]. During subsequent infections, the CRISPR system must be able to recognize and cleave the viral DNA (which contains the PAM) while avoiding the bacterial genome (where the spacer sequences lack PAMs) [17] [18]. The PAM sequence is thus not part of the guide RNA target but must be present adjacent to the target site in the DNA for successful recognition and cleavage by Cas nuclease [17].
From a structural perspective, PAM recognition occurs through specific interactions between the PAM DNA and particular domains of the Cas protein. For the commonly used Streptococcus pyogenes Cas9 (SpCas9), the PAM is recognized by a arginine-rich motif in the C-terminal domain of the protein [20]. This interaction triggers conformational changes that facilitate DNA unwinding and subsequent RNA-DNA hybridization [20]. The PAM sequence therefore serves as an essential binding signal that must be identified before Cas9 can proceed with checking for complementarity between the guide RNA and target DNA [17]. This sequential recognition mechanism—PAM identification followed by target verification—ensures both efficiency and specificity in DNA targeting.
PAM Sequences Across Different Cas Nucleases
Different Cas nucleases recognize distinct PAM sequences, which significantly influences their targeting ranges and applications. The canonical SpCas9 requires a 5'-NGG-3' PAM (where "N" can be any nucleotide base) immediately following the target sequence [17] [18]. However, the natural diversity of CRISPR systems and recent protein engineering efforts have yielded Cas variants with altered PAM specificities, substantially expanding the targetable genome [17] [21].
Table 1: PAM Sequences for Various CRISPR Nucleases
| CRISPR Nuclease | Organism/Source | PAM Sequence (5' to 3') | Notes |
|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | NGG | Canonical Cas9; most widely used [17] |
| SpCas9-NG | Engineered SpCas9 | NG | Expanded targeting range [21] |
| SpRY | Engineered SpCas9 | NRN > NYN | Near-PAMless variant [21] |
| Cas12a (Cpf1) | Lachnospiraceae bacterium | TTTV | Creates staggered ends [17] |
| SaCas9 | Staphylococcus aureus | NNGRRT or NNGRRN | Smaller size for viral delivery [17] |
| NmeCas9 | Neisseria meningitidis | NNNNGATT | Longer PAM; high specificity [17] |
| Cas12b | Alicyclobacillus acidiphilus | TTN | Thermostable variant [17] |
| Cas14 | Uncultivated archaea | T-rich (e.g., TTTA) | Targets single-stranded DNA [17] |
The diversity of PAM requirements across different Cas nucleases provides researchers with a toolbox of targeting options. For example, Cas12a (Cpf1) recognizes T-rich PAM sequences (TTTV, where V is A, C, or G) and creates staggered DNA ends rather than blunt cuts, which can be advantageous for certain applications [17]. Smaller Cas variants such as SaCas9 offer more compact coding sequences suitable for viral vector delivery while maintaining robust activity [17]. Recent engineering approaches have dramatically expanded PAM compatibilities, with variants like SpRY recognizing virtually all possible PAM sequences with efficiencies of NRN > NYN (where R is A/G and Y is C/T) [21]. This ongoing expansion of PAM diversity continues to broaden the targeting scope of CRISPR technologies.
Comparative Analysis of Genome Editing Technologies
Mechanism of Target Recognition
The three major genome editing technologies employ fundamentally different mechanisms for DNA recognition, which directly impacts their targeting flexibility, specificity, and ease of design:
CRISPR-Cas9: The CRISPR-Cas9 system relies on RNA-DNA complementarity for target recognition. A single guide RNA (sgRNA) of approximately 20 nucleotides directs the Cas9 nuclease to complementary genomic sequences [14]. Critical to this recognition is the requirement for a specific PAM sequence adjacent to the target site [17]. The Cas9 protein first scans DNA for the appropriate PAM sequence; upon identifying a PAM, it unwinds the adjacent DNA to allow hybridization with the guide RNA [20]. If sufficient complementarity exists, particularly in the "seed" region proximal to the PAM, Cas9 introduces a double-strand break 3-4 nucleotides upstream of the PAM [17].
TALENs: Transcription Activator-Like Effector Nucleases utilize a pair of customizable protein-DNA interactions for targeting [16]. Each TALEN consists of a DNA-binding domain derived from plant pathogenic bacteria fused to the FokI nuclease domain [19]. The DNA-binding domain comprises tandem repeats of 33-35 amino acids, with each repeat recognizing a single nucleotide through two hypervariable residues (Repeat Variable Diresidues or RVDs) [19] [16]. The RVD code follows a predictable pattern: NI recognizes adenine, HD recognizes cytosine, NG recognizes thymine, and NN recognizes guanine [16]. TALENs function as pairs binding to opposite DNA strands with a spacer sequence between them, enabling FokI dimerization and DNA cleavage [16].
ZFNs: Zinc Finger Nucleases also operate as pairs utilizing protein-DNA interactions but employ a different recognition code. Each zinc finger domain recognizes approximately 3 base pairs of DNA [19] [14]. Arrays of multiple zinc fingers (typically 3-6) are fused to the FokI nuclease domain to create target specificity [19]. Like TALENs, ZFNs must bind as pairs to opposite DNA strands with a spacer between them to facilitate FokI dimerization and DNA cleavage [14]. However, zinc finger arrays exhibit context-dependent effects where the recognition specificity of individual fingers can be influenced by neighboring fingers, making ZFN design more challenging [19].
Figure 1: Comparative mechanisms of target recognition by CRISPR, TALENs, and ZFNs
Targeting Flexibility and Limitations
The PAM requirement of CRISPR systems imposes a significant constraint on targeting flexibility compared to TALENs and ZFNs. While CRISPR guide RNAs can be easily designed to target virtually any sequence preceding an appropriate PAM, the absolute dependence on this short motif means that certain genomic regions may be inaccessible with standard Cas variants [17] [18]. For example, the canonical SpCas9 requiring 5'-NGG-3' PAM occurs approximately every 8-12 base pairs in random DNA sequence, but in practice, the distribution is non-random and some genomic regions may lack suitable PAMs for targeting specific nucleotides of interest [17].
In contrast, TALENs and ZFNs offer greater theoretical targeting flexibility since they do not operate under PAM constraints [16]. TALENs can potentially target any sequence with appropriate design, as the recognition is based on a straightforward one-to-one protein-DNA binding code [19]. Similarly, ZFNs can be designed to recognize a wide range of sequences, though the context-dependency of zinc finger arrays makes some targets more challenging than others [19]. However, both TALENs and ZFNs require designing and delivering pairs of proteins for each target, which complicates experimental design and implementation, particularly for multiplexed applications [16].
Table 2: Targeting Flexibility Comparison of Genome Editing Technologies
| Feature | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Recognition Mechanism | RNA-DNA hybridization | Protein-DNA (1bp/RVD) | Protein-DNA (3bp/zinc finger) |
| Sequence Constraint | Requires PAM sequence (e.g., NGG for SpCas9) | No PAM requirement | No PAM requirement |
| Targeting Density | Limited by PAM frequency | Potentially any sequence | Limited by zinc finger availability |
| Multiplexing Capacity | High (multiple gRNAs) | Low (pairs of large proteins) | Low (pairs of proteins) |
| Ease of Design | Simple (design sgRNA sequence) | Moderate (design TALE arrays) | Complex (context-dependent zinc fingers) |
| Development Time | Days | Days to weeks | Weeks to months [10] |
Recent advances in Cas engineering have substantially addressed CRISPR's PAM limitation. Through directed evolution and structure-guided engineering, researchers have developed Cas9 variants with altered PAM specificities [21]. Notable examples include xCas9 and SpCas9-NG variants that recognize NG PAMs, and SpRY that approaches PAM-less behavior [21]. A 2025 study published in Nature combined high-throughput protein engineering with machine learning to generate nearly 1,000 engineered SpCas9 enzymes with diverse PAM requirements, enabling "bespoke editors" for specific targets [21]. This evolving landscape continues to narrow the targeting flexibility gap between CRISPR and earlier technologies.
Experimental Evidence: Efficiency and Specificity Comparisons
Editing Efficiency and Pattern Analysis
Direct comparative studies reveal significant differences in editing efficiencies between CRISPR-Cas9 and TALENs. In a 2016 study comparing these technologies for editing an integrated EGFP gene in HEK293FT cells, researchers found that paired Cas9 nucleases induced targeted genomic deletion more efficiently and precisely than two TALEN pairs [15]. However, when concurrently supplied with a plasmid template spanning two double-strand breaks within EGFP, TALENs stimulated homology-directed repair more efficiently than CRISPR/Cas9 and caused fewer targeted genomic deletions [15]. This suggests that the choice of genome editing tool should be determined by the desired genomic outcome, with CRISPR potentially superior for gene knockouts through NHEJ, and TALENs possibly more efficient for precise gene insertion through HDR.
A 2018 study specifically compared editing patterns and efficiencies at the beginning of the CCR5 gene, an important therapeutic target for HIV treatment [22]. The results demonstrated that CRISPR-Cas9 mediated the sorting of cells that contained 4.8 times more gene editing than TALEN-transfected cells [22]. This substantial difference in editing efficiency highlights one of CRISPR's significant advantages for therapeutic applications where high editing rates are critical. The study employed detailed DNA sequencing to analyze editing patterns, revealing that both technologies produced diverse indels at the target site, but with different distribution profiles [22].
Table 3: Quantitative Comparison of Editing Efficiencies from Experimental Studies
| Study | Target | Cell Type | Editing Efficiency (CRISPR) | Editing Efficiency (TALEN) | Notes |
|---|---|---|---|---|---|
| He et al. (2016) [15] | EGFP | HEK293FT | High (paired nucleases) | Moderate | CRISPR more efficient for genomic deletions |
| Wang et al. (2018) [22] | CCR5 | Human cells | 4.8x higher | Baseline | CRISPR significantly more efficient |
| GeneCopoeia Comparison [16] | Various | Multiple | >70% indel formation | ~33% indel formation | CRISPR generally higher efficiency |
Off-Target Effects and Specificity
Specificity remains a critical consideration for all genome editing technologies, particularly for therapeutic applications. CRISPR-Cas9 has been reported to tolerate some mismatches between the guide RNA and target DNA, especially in the 5' region distal from the PAM, potentially leading to off-target effects at sites with similar sequences [16]. One study noted that sgRNAs can tolerate up to five mismatches with unintended target sites, though the extent of off-target activity varies significantly between different sgRNAs and cell types [16].
TALENs generally demonstrate higher specificity with fewer off-target effects, attributed to their requirement for longer recognition sequences and the obligate dimerization of FokI nuclease domains [16] [22]. The need for two TALEN proteins to bind in close proximity at the target site significantly reduces the probability of off-target cleavage. Similarly, ZFNs benefit from the FokI dimerization requirement, though their off-target profiles can be more variable due to the challenges in designing highly specific zinc finger arrays [19].
Several strategies have been developed to improve CRISPR specificity, including:
- Truncated sgRNAs (17-18 nucleotides instead of 20) that reduce off-target cleavage while maintaining on-target activity [16] [22]
- Paired nickases using Cas9 mutants that make single-strand breaks rather than double-strand breaks, requiring two adjacent sgRNAs for cutting [16]
- High-fidelity Cas9 variants (e.g., SpCas9-HF1, eSpCas9) with engineered mutations that reduce non-specific interactions with DNA [14]
- RNA-guided FokI nucleases (RFNs) that fuse catalytically inactive Cas9 to FokI, reinstating the dimerization requirement for activity [16]
Figure 2: Strategies to address off-target effects in genome editing technologies
Experimental Protocols for Technology Comparison
Side-by-Side Editing Assessment Protocol
To directly compare the editing efficiencies of CRISPR-Cas9 and TALENs, researchers can implement the following experimental protocol adapted from published comparative studies [15] [22]:
1. Target Selection and Editor Design:
- Select a target gene of interest with known sequence and function (e.g., EGFP or CCR5 as in published studies)
- For CRISPR-Cas9: Design sgRNAs targeting the region of interest using established tools, ensuring each target is followed by an appropriate PAM (5'-NGG-3' for SpCas9). Cloning involves synthesizing oligonucleotides corresponding to the sgRNA sequence and inserting them into an appropriate expression plasmid containing the Cas9 coding sequence [15] [22]
- For TALENs: Design TALEN pairs targeting the same region using online tools such as ZiFiT Targeter. Each TALEN should target 15-20 bp with a spacer of 14-20 bp between them. Assembly typically employs Golden Gate cloning with modular TALE repeat arrays [15]
2. Vector Construction:
- CRISPR-Cas9: Clone sgRNA sequences into a Cas9 expression plasmid (e.g., Addgene #41815). Many CRISPR plasmids incorporate fluorescent markers (e.g., GFP) or antibiotic resistance genes for selection [22]
- TALENs: Assemble TALEN pairs using established protocols such as the Golden Gate method. Co-transfect with a reporter plasmid that expresses RFP upon transfection and GFP only upon successful TALEN cleavage and repair-mediated frame correction [22]
3. Cell Transfection and Selection:
- Culture appropriate cell lines (e.g., HEK293FT) and seed in 24-well plates at 1.5×10^5 cells/well
- Transfect using recommended reagents (e.g., X-tremeGENE HP):
- For CRISPR: 0.3 μg Cas9 plasmid + 0.1 μg of each sgRNA plasmid (for paired nucleases)
- For TALENs: 0.1 μg of each TALEN plasmid + 0.1 μg reporter plasmid [15]
- Include untransfected controls and single-transfected controls where appropriate
4. Analysis of Editing Efficiency:
- Harvest cells 3 days post-transfection for initial assessment of editing
- Extract genomic DNA using commercial kits (e.g., DNeasy Blood & Tissue Kit)
- Amplify target region by PCR and assess editing using:
5. Functional Assessment:
- For reporter genes like EGFP, use flow cytometry to quantify functional knockout rates 7 days post-transfection [15]
- For endogenous genes, perform functional assays appropriate to the target (e.g., antibody staining for surface receptors like CCR5) [22]
Assessment of Off-Target Effects
Evaluating the specificity of editing technologies is crucial for applications requiring high precision:
1. In Silico Prediction of Off-Target Sites:
- For CRISPR: Identify potential off-target sites using tools that search for genomic sequences with similarity to the sgRNA target, allowing for mismatches, especially in the 5' region
- For TALENs: Identify sites with similarity to the TALEN binding sites, considering the degeneracy of RVD codes
2. Experimental Detection Methods:
- GUIDE-seq: Genome-wide, unbiased identification of DSBs enabled by sequencing uses integration of oligonucleotides into DSB sites to identify both on-target and off-target cleavage sites [18]
- * targeted sequencing*: Deep sequencing of predicted off-target sites to quantify editing frequencies
- Whole-genome sequencing for comprehensive identification of off-target effects in clonal populations
Successful implementation of genome editing technologies requires specific reagent systems and tools. The following table outlines key solutions for researchers embarking on comparative studies of CRISPR and TALEN technologies:
Table 4: Essential Research Reagents for Genome Editing Studies
| Reagent/Resource | Function | CRISPR-Specific Notes | TALEN-Specific Notes |
|---|---|---|---|
| Cas9 Expression Plasmid | Expresses Cas9 nuclease | Codon-optimized versions available for different species; high-fidelity variants reduce off-target effects | Not applicable |
| TALEN Expression Vectors | Expresses TALEN proteins | Not applicable | Typically require pair of plasmids for left and right TALENs |
| sgRNA Cloning Vector | Template for sgRNA expression | U6 promoter drives expression; requires insertion of 20nt target sequence | Not applicable |
| Reporter Plasmid | Assesses editing efficiency | Often incorporated into Cas9 vector (e.g., T2A-GFP) | Separate plasmid expressing RFP/GFP upon successful editing [22] |
| Delivery Reagents | Introduces editing components into cells | Lipid-based transfection, electroporation, or viral delivery | Similar delivery methods, but larger payload size for TALENs |
| Target Validation Tools | Confirms editing at target site | Surveyor/T7E1 assay, tracking of indels by decomposition (TIDE), next-generation sequencing | Same validation methods applicable |
| Off-Target Assessment | Identifies unintended edits | GUIDE-seq, CIRCLE-seq, targeted sequencing | Similar approaches but generally fewer off-target concerns |
The selection of appropriate reagents significantly influences editing outcomes. For CRISPR systems, the choice of Cas9 variant (wild-type, high-fidelity, nickase) determines both targeting range and specificity [14]. For TALENs, the design of RVD arrays and spacer length between binding sites affects activity and specificity [16]. Recent commercial sources provide pre-validated editors for common targets, reducing development time and improving success rates, particularly for researchers new to genome editing.
The critical role of PAM sequences in CRISPR technology represents both a constraint and an opportunity for genome engineering applications. While the PAM requirement limits the theoretical targeting space compared to TALENs and ZFNs, ongoing engineering efforts are rapidly expanding this boundary. The development of PAM-flexible Cas variants through directed evolution and structure-based design continues to increase the targetable genome [21]. A 2025 study demonstrated the power of combining high-throughput protein engineering with machine learning to generate bespoke Cas9 variants with customized PAM specificities, potentially enabling allele-selective editing for therapeutic applications [21].
The choice between CRISPR, TALENs, and ZFNs depends heavily on the specific research or therapeutic context. CRISPR-Cas9 offers unparalleled ease of design, high efficiency, and straightforward multiplexing capabilities, making it ideal for most research applications, particularly gene knockouts [15] [22]. TALENs provide high specificity with minimal off-target effects and no PAM constraints, advantageous for applications requiring precise editing in sensitive genomic contexts [16]. ZFNs, while more challenging to design, offer compact coding sequences and established safety profiles that have enabled their use in clinical trials [19].
Future developments in genome editing will likely focus on expanding targeting scope, improving specificity, and enhancing editing precision. For CRISPR systems, continued engineering of Cas variants with novel PAM specificities will further close the gap in targeting flexibility [21]. The integration of machine learning approaches to predict both on-target efficiency and off-target effects for specific guide RNAs will improve experimental design and outcomes [21]. As these technologies mature, the critical role of PAM sequences in determining targetable sites will continue to evolve, potentially leading to truly PAM-independent editing systems that maintain high specificity while offering complete targeting freedom across the genome.
Inherent FokI Dimerization in TALENs/ZFNs as a Natural Specificity Check
The quest for precision in genome editing has positioned engineered nucleases as powerful tools for research and therapy. Within this landscape, the inherent structural requirement for FokI nuclease dimerization in Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs) acts as a critical, built-in mechanism that enhances target specificity. This biological safeguard provides a compelling contrast to the mechanism of the more recent CRISPR-Cas9 system. This guide objectively compares the experimental performance of these platforms, focusing on how the FokI dimerization checkpoint influences editing accuracy, supported by direct experimental data and protocols relevant to drug development professionals.
Mechanism of Action: A Tale of Two Architectures
The core difference in specificity control between platform types lies in their fundamental architecture for DNA recognition and cleavage.
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| DNA Recognition Mechanism | Protein-DNA interaction [23] | Protein-DNA interaction [23] | RNA-DNA base pairing [23] [8] |
| Recognition Site Length | 9-18 bp [23] [9] | 30-40 bp [23] | 22 bp + PAM sequence [23] |
| Cleavage Domain | FokI nuclease [23] [9] | FokI nuclease [23] | Cas9 nuclease [23] |
| Cleavage Requirement | Obligate FokI Dimerization [23] [9] | Obligate FokI Dimerization [23] | Single Protein with Dual Nuclease Activity [23] |
The following diagram illustrates the critical mechanistic difference: the requirement for two independent binding events in ZFNs and TALENs versus the single-guide system of CRISPR-Cas9.
Experimental Evidence: Quantifying the Specificity Advantage
The theoretical specificity advantage of FokI dimerization has been validated in numerous direct comparative studies. The data below summarize key experimental findings measuring on-target efficiency versus off-target activity.
Table 1: Experimental Data from Direct Nuclease Comparisons. This table synthesizes data from multiple studies quantifying gene targeting efficiency and off-target effects [24] [25] [8].
| Nuclease Platform | Experimental System | Reported On-Target Efficiency | Reported Off-Target Effects | Key Finding |
|---|---|---|---|---|
| HBB-TALENs | EGFP Reporter Assay in 293T cells [24] | 0.35% HR Frequency [24] | Detected on HBD target (2.4% activity) [24] | Demonstrated that longer recognition sites do not guarantee higher specificity. |
| HBB-ZFNs | EGFP Reporter Assay in 293T cells [24] | ~0.2% HR Frequency [24] | Low level on HBE and HBG sequences [24] | Lower efficiency than more recently developed ZFNs (ZFA pair). |
| HBB-ZFA (Improved ZFN) | EGFP Reporter Assay in 293T cells [24] | ~0.9% HR Frequency [24] | Not detected on related HBD, HBE, HBG sequences [24] | Illustrates that well-designed ZFNs can achieve high specificity. |
| HBB Hybrid (ZFA-L + TALEN1-R) | EGFP Reporter Assay in 293T & human iPS cells [24] | 2.8% HR Frequency (8-fold higher than TALEN1 pair) [24] | Not detected on related HBD, HBE, HBG sequences [24] | Hybrid nuclease showed highest efficiency and no detected off-target activity. |
| Anti-HBV TALENs (2nd Gen Heterodimer) | HBV-transfected Huh7 cells [25] | Similar silencing efficacy to 1st-gen TALENs [25] | Improved specificity in mouse model of HBV replication [25] | Engineering obligate heterodimers maintained efficacy while improving specificity. |
Case Study: Enhanced Specificity with Obligate Heterodimers
A 2021 study on improving TALENs for hepatitis B virus (HBV) therapy directly engineered the FokI domain to create "obligate heterodimeric TALENs" [25]. This approach uses second- and third-generation FokI nuclease domains that are only active when two different TALEN monomers (left and right) dimerize. This prevents cleavage from homodimers (two identical subunits), which could form at off-target sites with similar, but not identical, sequences [25]. The results confirmed that these modified TALENs maintained high activity against viral DNA while demonstrating an improved specificity profile in an in vivo model, showcasing a direct engineering application of the dimerization principle [25].
Detailed Experimental Protocol: Assessing Nuclease Specificity
To empower researchers in validating these findings, below is a detailed methodology for a key experiment used to quantify nuclease activity and off-target effects—the EGFP Reporter Assay, adapted from the studies cited [24].
EGFP Reporter Assay for Homologous Recombination (HR) Frequency
Objective: To quantitatively measure the frequency of precise, nuclease-induced gene correction via Homologous recombination in a cell population.
Principle: A cell line harbors a chromosomally integrated, non-functional Enhanced Green Fluorescent Protein (EGFP) gene, disrupted by a stop codon and the target DNA sequence of interest. Transfection with nuclease plasmids and a donor DNA template (tGFP) enables HR. Successful correction restores a functional EGFP gene, and cells expressing EGFP are quantified via flow cytometry [24].
Materials & Reagents:
- Stable Reporter Cell Line: e.g., HEK293T or other relevant cell line with integrated, disrupted EGFP gene containing the target site.
- Nuclease Expression Vectors: Plasmids expressing the left and right monomers of the TALEN or ZFN pair, or a plasmid expressing Cas9 and the specific sgRNA.
- Donor DNA Template: A non-expressing "tGFP" donor DNA plasmid for HR, containing the corrected EGFP sequence with the target site removed/mutated.
- Transfection Reagent: e.g., Lipofectamine 3000.
- Flow Cytometer: For detecting and quantifying EGFP-positive cells.
Procedure:
- Cell Seeding: Seed the stable reporter cells in a multi-well plate (e.g., 6-well) at 50% confluency one day before transfection.
- Transfection: Co-transfect the cells with:
- 1 µg of each nuclease monomer plasmid (for ZFNs/TALENs) or 2 µg of CRISPR-Cas9/sgRNA plasmid.
- 300 ng of the pCH-9/3091 (or similar) target plasmid if required.
- 200 ng of the tGFP donor DNA template.
- 200 ng of a transfection control plasmid (e.g., pCI-neo eGFP).
- Use a mock transfection control (e.g., pUC118 plasmid) in place of nuclease plasmids.
- Incubation: Incubate the cells for 48-72 hours post-transfection.
- Analysis:
- Visualize transfection efficiency by fluorescence microscopy using the control GFP.
- Harvest cells and resuspend in buffer for flow cytometry.
- Analyze at least 100,000 events per sample to detect EGFP-positive cells.
- Calculate HR Frequency: (Number of EGFP-positive cells / Total number of cells analyzed) × 100%.
The Scientist's Toolkit: Essential Reagents for Nuclease Studies
Table 2: Key Research Reagents for Genome Editing Experiments. This table catalogs essential materials used in the featured studies and their critical functions in nuclease research [24] [25].
| Reagent / Solution | Function / Description | Example Use Case |
|---|---|---|
| TALEN/ZFN Expression Vectors (pVAX, etc.) | Plasmid backbones (e.g., with CMV promoter) for expressing nuclease monomers in mammalian cells [25]. | Delivery of TALEN or ZFN pairs into target cells for gene editing [24]. |
| FokI Nuclease Domain (Wild-type & Obligate Heterodimer Mutants) | The catalytic domain that cleaves DNA; engineered variants require two different monomers to form an active complex [25] [9]. | Enhancing specificity by preventing homodimer formation and off-target cleavage [25]. |
| EGFP Reporter Cell Line | Engineered cell line with a disrupted EGFP gene that can be restored via nuclease-driven HR [24]. | Quantitative measurement of homologous recombination efficiency and functional nuclease activity [24]. |
| Donor DNA Template (ssODN or dsDNA) | A homologous repair template containing the desired genetic modification flanked by homology arms [9]. | Enabling precise gene knock-in or correction via the HDR pathway [24]. |
| Lipofectamine 3000 | A common liposomal transfection reagent for delivering nucleic acids into cultured cells. | Transient transfection of nuclease plasmids and donor DNA into adherent cell lines [25]. |
| Surveyor Nuclease Assay (or T7E1 Assay) | A mismatch-specific endonuclease used to detect nuclease-induced indel mutations at the target site. | Validation of on-target editing efficiency and initial screening for potential off-target sites [25]. |
The inherent need for FokI dimerization in ZFNs and TALENs provides a fundamental, protein-based checkpoint that is absent in the standard single-guide RNA-driven CRISPR-Cas9 system. Experimental data consistently demonstrates that this architecture can result in lower off-target effects, a critical consideration for therapeutic applications. While CRISPR offers unparalleled ease of use and flexibility, the continued development and application of ZFNs and TALENs—especially with advanced FokI engineering—remain vital for projects where the highest possible specificity is non-negotiable. The choice of platform should therefore be guided by a balanced consideration of the project's requirements for ease of design, efficiency, and most importantly, precision.
From Bench to Bedside: Application-Based Selection of Editing Tools
Gene editing has ushered in a transformative era for treating monogenic diseases, moving from theoretical concept to clinical reality. The landscape of programmable nucleases has been historically dominated by zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), which require complex protein engineering for each new DNA target [26]. The advent of CRISPR-Cas9 has revolutionized the field by introducing an RNA-guided system, drastically simplifying design and enabling rapid deployment across diverse applications [8]. This guide provides an objective comparison of these platforms, focusing on their therapeutic performance in three landmark diseases: sickle cell disease (SCD), transfusion-dependent beta thalassemia (TBT), and hereditary transthyretin amyloidosis (hATTR). We present structured experimental data, detailed methodologies, and analytical visualizations to illustrate the factors behind CRISPR's emerging dominance in the clinical arena.
Clinical Trial Performance: Quantitative Outcomes Comparison
The most compelling evidence for a therapeutic technology lies in its clinical trial data. The following tables summarize key efficacy and safety outcomes for CRISPR-based therapies across the three target diseases, highlighting the direct clinical results that have propelled CRISPR to the forefront.
Table 1: Efficacy Outcomes from Pivotal CRISPR Clinical Trials
| Disease | Therapy / Trial | Primary Efficacy Endpoint | Result | Patient Population |
|---|---|---|---|---|
| Sickle Cell Disease (SCD) | Casgevy (ex vivo CRISPR-Cas9) [27] | Freedom from severe vaso-occlusive crises (VOCs) for ≥12 consecutive months | 93.5% (29 of 31 evaluable patients) | Patients 12+, with history of recurrent VOCs |
| Transfusion-Dependent Beta Thalassemia (TBT) | Casgevy (ex vivo CRISPR-Cas9) [28] | Freedom from regular blood transfusions for ≥12 consecutive months | 94% (c. 36 of 38 evaluable patients, based on IGI report) | Patients 12+, requiring regular transfusions |
| hATTR Amyloidosis | NTLA-2001 (in vivo CRISPR-Cas9) [28] [29] | Reduction in serum transthyretin (TTR) protein | ~90% reduction sustained at 28 days and through 2-year follow-up | Patients with hATTR with polyneuropathy or cardiomyopathy |
Table 2: Safety and Delivery Profile Comparison
| Therapy | Delivery Method | Common Adverse Events | Notable Risks | Therapeutic Dosing |
|---|---|---|---|---|
| Casgevy (SCD/TBT) [27] | Ex vivo (CD34+ HSPCs) | Low platelets/white blood cells, mouth sores, nausea, febrile neutropenia | Requires myeloablative conditioning | One-time, single-dose infusion |
| NTLA-2001 (hATTR) [28] [30] | In vivo (Systemic LNP) | Mild or moderate infusion-related reactions | No evidence of declining effect over 2 years | Single-dose IV infusion; potential for redosing |
Platform Comparison: CRISPR vs. TALENs vs. ZFNs
While all three nuclease platforms can create targeted double-strand breaks in DNA, their fundamental mechanisms, design processes, and practical implementation differ significantly. The following table provides a structured comparison of their core characteristics, while the diagram illustrates the fundamental mechanistic differences that underpin these practical distinctions.
Table 3: Core Technology Comparison of Programmable Nucleases
| Feature | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Targeting Mechanism | RNA-guided (gRNA) via Watson-Crick base pairing [26] | Protein-based (TALE repeats) | Protein-based (Zinc finger domains) |
| Nuclease Component | Cas9 (HNH & RuvC domains) [26] | FokI dimer [8] [26] | FokI dimer [8] [26] |
| Design & Development | Simple, rapid gRNA design (days) [8] | Labor-intensive TALE repeat assembly (weeks-months) [8] [26] | Complex zinc finger engineering (weeks-months) [8] [26] |
| Target Specificity | Moderate to high; subject to off-target effects [8] | High; reduced off-target risks [8] | High; proven precision in clinical edits [8] |
| Multiplexing Capacity | High (multiple gRNAs simultaneously) [8] | Limited [8] | Very limited [8] |
| Cost Efficiency | Low [8] | High [8] | High [8] |
Diagram 1: Mechanisms of Major Gene-Editing Platforms. CRISPR-Cas9 uses a guide RNA for DNA targeting, while TALENs and ZFNs rely on protein-DNA binding and FokI dimerization for cleavage.
Detailed Experimental Protocols
Ex Vivo Editing for SCD: Casgevy (CTX001)
The Casgevy therapy for SCD is a prime example of ex vivo CRISPR editing. The protocol involves isolating a patient's own hematopoietic stem and progenitor cells (HSPCs), genetically modifying them outside the body, and then reinfusing them to establish a new, healthy blood system [31] [27].
- HSPC Collection & Isolation: CD34+ HSPCs are collected from the patient via apheresis after mobilization from the bone marrow [31] [27].
- Ex Vivo Editing:
- Myeloablative Conditioning: The patient receives busulfan chemotherapy to clear the bone marrow niche and make space for the engraftment of the edited cells [27].
- Reinfusion & Engraftment: The CRISPR-edited CD34+ cells are infused back into the patient, where they engraft in the bone marrow [27]. The disruption of BCL11A leads to sustained elevation of HbF, which prevents the sickling of red blood cells, thereby ameliorating the disease [31] [27].
In Vivo Editing for hATTR: NTLA-2001
The NTLA-2001 trial for hATTR amyloidosis represents a breakthrough for in vivo CRISPR therapy, where editing occurs directly inside the patient's body [28] [30].
- Vector Design & Formulation:
- The CRISPR-Cas9 system is packaged into lipid nanoparticles (LNPs) [28]. These are tiny fat particles that protect the editing machinery and facilitate its delivery to target cells.
- The Cas9 component used is a compact Staphylococcus aureus Cas9 (SaCas9) or other variant that fits within the LNP's packaging constraints [32].
- Systemic Administration: The LNPs are administered to the patient via a single intravenous (IV) infusion [28] [30].
- Hepatocyte Targeting & Gene Knockout: The LNPs naturally accumulate in the liver. Once inside hepatocytes, the CRISPR system is released and travels to the nucleus [28]. The guide RNA directs Cas9 to introduce a double-strand break in the TTR gene, disrupting its sequence and permanently reducing the production of the misfolded TTR protein responsible for the disease [29] [30].
Diagram 2: Ex Vivo vs. In Vivo CRISPR Therapeutic Workflows. Ex vivo editing modifies patient cells outside the body, while in vivo editing delivers CRISPR directly into the patient's cells.
The Scientist's Toolkit: Essential Research Reagents
Successful translation of CRISPR therapies from bench to bedside relies on a suite of critical reagents and tools. The following table details key solutions used in the development and manufacturing of these advanced therapies.
Table 4: Key Research Reagent Solutions for CRISPR Therapy Development
| Reagent / Solution | Function | Example Use in Featured Trials |
|---|---|---|
| CRISPR Ribonucleoprotein (RNP) | Complex of Cas9 protein and guide RNA; enables efficient, transient editing with reduced off-target effects [31]. | Direct delivery into CD34+ HSPCs for ex vivo editing in Casgevy [31]. |
| Lipid Nanoparticles (LNPs) | Non-viral delivery vector for in vivo administration; protects CRISPR components and facilitates cellular uptake [28]. | Systemic delivery of CRISPR-Cas9 for NTLA-2001 (hATTR) [28]. |
| CD34+ HSPC Culture Media | Specialized media formulations that maintain stem cell potency and viability during ex vivo manipulation [31]. | Expansion and maintenance of patient-derived HSPCs during the Casgevy manufacturing process [31]. |
| Adeno-Associated Virus (AAV) | Viral vector often used as a donor template for HDR; limited packaging capacity but high efficiency [32]. | Preclinical delivery of smaller Cas9 orthologs (e.g., Nme2Cas9, SaCas9) for in vivo editing [32]. |
| Electroporation Systems | Instrumentation that uses electrical pulses to create transient pores in cell membranes for intracellular delivery of editing components [31]. | Introduction of CRISPR RNP into hard-to-transfect primary CD34+ HSPCs in ex vivo protocols [31]. |
The accumulation of clinical data from trials for SCD, TBT, and hATTR provides compelling evidence of CRISPR's therapeutic dominance. Its success is rooted in a powerful combination of high clinical efficacy, demonstrated by durable responses in a majority of patients, and a versatile and streamlined platform that supports both ex vivo and in vivo approaches. While traditional methods like ZFNs and TALENs maintain relevance for niche applications requiring their validated high specificity, the ease of design, cost-effectiveness, and rapid iteration of CRISPR have accelerated its clinical translation and expanded the scope of treatable diseases. As delivery technologies, particularly LNPs, continue to advance, the potential of in vivo CRISPR editing promises to broaden this therapeutic landscape even further.
The advent of programmable nucleases has ushered in a transformative era for molecular biology, enabling precise modifications to DNA sequences across a diverse array of organisms. Among the most prominent tools in this arena are Zinc-Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the CRISPR/Cas9 system [8] [33]. While the CRISPR/Cas9 system has gained widespread popularity due to its ease of design and cost-effectiveness, TALENs have carved out a critical and enduring niche in applications demanding the utmost precision, particularly in mitochondrial DNA editing and therapeutic development where specificity is paramount [33] [34].
The fundamental difference between these platforms lies in their mechanism of target recognition. CRISPR/Cas9 is an RNA-guided system, where a short guide RNA (gRNA) directs the Cas9 nuclease to a complementary DNA sequence [16]. In contrast, TALENs utilize a protein-based recognition system; each TALEN is composed of a series of modular, 33-35 amino acid repeats that each recognizes a single DNA base pair through two critical amino acids known as the Repeat Variable Diresidue (RVD) [33] [16]. This structural distinction underpins the unique strengths and weaknesses of each platform.
Key Comparisons: TALENs vs. CRISPR-Cas9
Table 1: Fundamental Characteristics of TALENs and CRISPR/Cas9
| Feature | TALEN | CRISPR/Cas9 |
|---|---|---|
| Recognition Type | DNA-Protein | DNA-RNA |
| Target Site Length | 30-36 bp | ~23 bp |
| Nuclease | FokI (requires dimerization) | Cas9 (functions as a monomer) |
| Off-Target Activity | Low | Moderate to High |
| Design & Assembly | Labor-intensive protein engineering | Simple RNA programming |
| Target Range | Virtually unlimited | Limited by PAM sequence (e.g., 5'-NGG-3') |
| Methylation Sensitivity | Yes (sensitive to CpG methylation) | No |
| Mitochondrial Genome Editing | Straightforward (with MTS) | Extremely challenging |
Table 2: Performance Comparison in Key Applications
| Application | TALENs Performance | CRISPR/Cas9 Performance |
|---|---|---|
| Specificity (Off-Target Effects) | High specificity; long target site (≈36 bp) is statistically unique in the genome [33] [16]. | Moderate specificity; gRNA can tolerate mismatches, leading to more off-target effects [33] [16]. |
| Mitochondrial DNA Editing | Highly effective; protein-based editors are easily targeted to mitochondria [35] [34]. | Inefficient; no robust mechanism for importing gRNA into mitochondria [34] [36]. |
| Therapeutic Precision | Preferred for clinical applications requiring validated, high-specificity edits [8] [33]. | Concerns over off-target mutagenesis and immune responses to Cas9 can limit therapeutic use [8] [33]. |
| Efficiency of Indel Formation | High (e.g., ~33% shown in iPSCs) [16]. | Very High (can exceed 70%) [16]. |
| Multiplexing Capability | Limited; challenging protein engineering. | Excellent; multiple gRNAs can be used simultaneously [8]. |
The Case for TALENs in Mitochondrial Genome Editing
The Mitochondrial Challenge and the TALEN Solution
Mitochondrial diseases are often caused by heteroplasmic mutations in mitochondrial DNA (mtDNA), where a mixture of wild-type and mutant mtDNA molecules coexist within a cell [36]. The severity of disease is linked to the proportion of mutant mtDNA, and a key therapeutic strategy is to shift this heteroplasmy ratio toward the wild-type by selectively reducing mutant mtDNA [36]. However, editing the mitochondrial genome presents unique hurdles. Mitochondria have a double membrane and lack efficient mechanisms for importing foreign RNAs, which critically limits the utility of the RNA-guided CRISPR/Cas9 system [34] [36].
TALENs overcome this fundamental barrier. Because they function entirely as proteins, TALENs can be directed to the mitochondria by simply fusing them to a Mitochondrial Targeting Sequence (MTS). This has led to the development of powerful tools like mitoTALENs and, more recently, TALEN-based mitochondrial base editors such as DddA-derived Cytosine Base Editors (DdCBEs) [35] [34] [36]. These systems have demonstrated remarkable success in selectively degrading mutant mtDNA or directly correcting point mutations, thereby restoring cellular energy production and function.
Experimental Evidence and Protocol: Correcting the m.8993T>G Mutation
A seminal 2025 study showcased the therapeutic potential of TALEN-based mitochondrial base editing by correcting the pathogenic m.8993T>G mutation in the MT-ATP6 gene, which causes Neurogenic Muscle Weakness, Ataxia, and Retinitis Pigmentosa (NARP) [35].
- Objective: To reduce the high heteroplasmy level (80%) of the m.8993T>G mutation in patient-derived induced pluripotent stem cells (iPSCs) and restore mitochondrial function.
- Editor Design: A split-DddA cytosine base editor (DdCBE) was constructed. Each half was fused to:
- A COX8A Mitochondrial Targeting Sequence (MTS).
- A programmable TALE DNA-binding domain designed to recognize a 16 bp sequence flanking the MT-ATP6 position 8993.
- One half of the split bacterial deaminase DddA [35].
- Delivery & Workflow: The two editor halves were delivered into patient iPSCs via nucleofection. Cells were analyzed over a 30-day period for editing efficiency, off-target effects, and functional recovery [35].
- Results:
- Editing Efficiency: Achieved 35 ± 3% on-target C•G to T•A conversion at day 7, successfully reducing mutant heteroplasmy from 80% to 45% [35].
- Specificity: Demonstrated minimal off-target editing (<0.5%) at ten predicted off-target loci in the mitochondrial genome [35].
- Functional Rescue: Edited cells showed a 25% increase in basal oxygen consumption rate, a 50% improvement in ATP-linked respiration, and a 2.3-fold restoration of ATP synthase activity. Furthermore, neural differentiation was significantly enhanced [35].
The following diagram illustrates the experimental workflow and mechanism of action for the TALEN-based mitochondrial base editor in this study.
TALENs in High-Specificity Nuclear Genome Editing
Beyond mitochondrial applications, TALENs maintain a strong position in nuclear genome editing projects where minimizing off-target effects is the primary concern. Comprehensive specificity profiling studies have revealed that TALENs are generally less prone to off-target cleavage than first-generation CRISPR/Cas9 systems [37] [16].
A 2014 study used an in vitro selection method to profile the specificity of 30 unique TALENs against a library of over 10^12 potential off-target sequences [37]. The results demonstrated that TALENs are highly specific across their entire target sequence. The study also led to the engineering of a "Q3" TALEN variant, which exhibited a further 10-fold reduction in average off-target activity in human cells while maintaining robust on-target cleavage [37].
Furthermore, a direct comparative study on editing an integrated EGFP gene in HEK293FT cells found that the choice of editor should be determined by the desired outcome [15]. While paired Cas9 nucleases were more efficient at inducing targeted genomic deletions, TALENs stimulated Homology-Directed Repair (HDR) more efficiently and caused fewer unintended deletions when supplied with a plasmid repair template [15]. This makes TALENs a superior choice for applications requiring precise gene correction or knock-in.
The following diagram outlines the basic mechanism of action for TALENs in the nucleus, highlighting the source of their high specificity.
Essential Reagents and Tools for TALEN Research
Table 3: The Scientist's Toolkit for TALEN-based Experiments
| Reagent / Tool | Function | Example in Context |
|---|---|---|
| TALE Repeat Plasmids | Modular building blocks for assembling the DNA-binding domain. | Pre-designed RVD modules (e.g., NI for A, HD for C, NG for T, NN for G) are ligated to target a specific sequence [15] [16]. |
| FokI Nuclease Domain | The cleavage module that induces a double-strand break. | Typically used as an obligate heterodimer (e.g., ELD/KKR variants) to improve specificity and reduce homodimer off-target activity [37]. |
| Mitochondrial Targeting Sequence (MTS) | Directs the TALEN protein to the mitochondrial matrix. | A short peptide sequence (e.g., from COX8A) is fused to the N-terminus of the TALEN for mitoTALEN or DdCBE constructs [35] [36]. |
| DddA-derived Cytosine Deaminase | Catalyzes C•G to T•A base conversions without double-strand breaks. | Used in a split-half configuration fused to TALE arrays for mitochondrial base editing (DdCBE) [35]. |
| Delivery Vectors | Plasmid or mRNA constructs for delivering editors into cells. | For iPSCs, plasmids encoding editor halves are often delivered via nucleofection [35]. AAV delivery is challenging due to TALEN's large size [36]. |
In the broader thesis of CRISPR vs. TALENs vs. ZFNs, the evidence firmly establishes that while CRISPR/Cas9 is an unparalleled tool for high-throughput screening and versatile research applications, TALENs hold a definitive niche superiority in contexts where precision is non-negotiable [8] [33]. Their protein-based DNA recognition mechanism makes them uniquely suited for mitochondrial genome editing, a field where CRISPR struggles due to fundamental delivery constraints [34] [36]. Furthermore, their inherently high specificity and efficiency in stimulating homology-directed repair cement their value in therapeutic development and other high-stakes applications where off-target effects carry significant risk [15] [37]. As the field of genetic medicine advances, TALENs and their evolved derivatives will remain indispensable tools for achieving precise genetic corrections in both the nuclear and mitochondrial genomes.
The advent of programmable nucleases has revolutionized genetic engineering in both agriculture and metabolic engineering. Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the CRISPR-Cas system constitute the three foundational platforms for targeted genome editing [26]. These technologies enable precise modifications to plant genomes, from single nucleotide changes to large insertions or deletions, facilitating the development of crops with enhanced agronomic traits and medicinal plants with optimized metabolic profiles.
These tools have dramatically accelerated breeding processes. In agriculture, over 60% of new crop varieties in 2025 utilize CRISPR editing for enhanced yield and disease resistance, demonstrating its rapid adoption and transformative impact [38]. Similarly, in metabolic engineering, these technologies are crucial for manipulating the biosynthetic pathways of plant natural products (PNPs), which are primary sources of cosmetics, food additives, and pharmaceuticals [39]. The choice of editing platform depends on the specific application, required precision, and resources, each offering distinct advantages and limitations for modifying crops and medicinal plants.
Technical Comparison of Editing Platforms
The core mechanisms, advantages, and limitations of ZFNs, TALENs, and CRISPR systems differ significantly, influencing their application in plant science. The following table provides a structured comparison of their key technical characteristics.
Table 1: Technical Comparison of Genome Editing Platforms
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| DNA Recognition Mechanism | Protein-DNA interaction (Zinc finger domains) [40] | Protein-DNA interaction (TALE repeats) [40] | RNA-DNA hybridization (Guide RNA) [40] [26] |
| Target Specificity | 9–18 bp (per monomer; dimer required) [19] [26] | 30–40 bp (per monomer; dimer required) [40] | 20 bp guide sequence + PAM (e.g., 5'-NGG-3') [40] |
| Nuclease Component | FokI endonuclease domain [19] [40] | FokI endonuclease domain [19] [40] | Cas9 nuclease (or variants) [40] [26] |
| Ease of Design & Cloning | Challenging; zinc finger motifs affect neighbors, requiring extensive engineering [19] [40] | Relatively easy; modular TALE repeats with defined specificity [19] [40] | Very easy; requires only guide RNA synthesis/cloning [40] [8] |
| Multiplexing Potential | Low; challenging and costly to design multiple proteins [8] | Low; challenging and costly to design multiple proteins [8] | High; multiple guide RNAs can be used simultaneously [8] |
| Primary Applications in Plants | Early proof-of-concept edits; stable cell line generation [8] | Editing complex loci; secondary metabolite pathway engineering [41] [39] | High-throughput gene knockout, screening, crop trait improvement, metabolic pathway engineering [38] [39] |
Mechanism of Action and Key Differentiators
The editing platforms function through distinct mechanisms to create double-strand breaks (DSBs) in DNA, which are then repaired by the cell's own repair machinery.
ZFNs and TALENs: Both are fusion proteins that pair a customizable DNA-binding domain with the catalytic domain of the FokI endonuclease [19] [40]. The FokI domain must dimerize to become active, meaning two ZFN or TALEN monomers must bind opposite strands of the DNA target site in close proximity to create a DSB [40]. The key difference lies in their DNA-binding domains: ZFNs use arrays of zinc finger motifs, each recognizing a 3-base pair DNA sequence, while TALENs use TALE repeats, where each repeat recognizes a single nucleotide [40] [26]. This makes TALEN design more straightforward and flexible.
CRISPR-Cas9: This system uses a guide RNA (gRNA) that is complementary to the target DNA sequence to direct the Cas9 nuclease to the genomic locus [26]. The target site must be adjacent to a short DNA sequence known as the Protospacer Adjacent Motif (PAM), which is essential for Cas9 recognition [40] [26]. Upon binding, the Cas9 enzyme uses its HNH and RuvC nuclease domains to cleave both DNA strands, creating a DSB [26]. The reliance on RNA-DNA base pairing, rather than protein-DNA interaction, is the primary reason for CRISPR's ease of design and reprogramming.
The following diagram illustrates the fundamental mechanisms by which each platform recognizes its target DNA and induces a double-strand break.
Applications in Crop Engineering
Genome editing technologies are being deployed to address pressing agricultural challenges such as climate change, pest pressure, and global food security. The following table summarizes key applications and achievements of each platform in crop improvement.
Table 2: Applications of Editing Platforms in Crop Engineering
| Editing Platform | Target Crops | Engineered Trait | Key Outcome/Impact |
|---|---|---|---|
| CRISPR-Cas9 | Wheat, Rice | Climate Resilience (Drought, Heat, Flooding) | Up to 20% yield increase under stress; Enables cultivation on marginal lands [38]. |
| CRISPR-Cas9 | Cassava (Africa) | Viral Disease Resistance | Secures food staple for millions; Addresses pressing food security challenge [38]. |
| CRISPR-Cas9 | Tomato, Potato, Maize | Pest and Disease Resistance | Reduces chemical pesticide use; Up to 15% reduction in crop losses [38]. |
| CRISPR-Cas9 | Rice, Maize, Wheat | Nutritional Optimization (Biofortification) | Enhanced levels of Vitamin A, iron, zinc to combat malnutrition [38]. |
| TALENs | Various Cereals | Nitrogen Use Efficiency | Decreased synthetic fertilizer requirement by up to 30%; lowers greenhouse gas emissions [38]. |
| TALENs & ZFNs | Barley, Sugar Beet | Disease Resistance, Lower Water Usage | Development of mildew-resistant barley and more water-use efficient sugar beet [42]. |
Trait Development and Workflow
The development of edited crops follows a structured pipeline from target identification to field trials. CRISPR dominates this space due to its efficiency and multiplexing capabilities. A prime example is the development of climate-resilient crops. Gene edits can enhance root architecture in wheat and rice, allowing them to thrive in conditions of water scarcity [38]. Similarly, CRISPR is used to create pest-resistant varieties of tomato, potato, and maize by introducing built-in resistance to common pests and pathogens, thereby reducing reliance on chemical pesticides [38].
The workflow often begins with AI-driven precision breeding, where AI-powered platforms analyze genomic data to predict gene functions and identify optimal CRISPR targets for complex traits like heat tolerance [42]. This synergy between digital and molecular tools is accelerating the development of crops adapted to changing environments.
Applications in Medicinal Plant and Metabolic Engineering
In medicinal plants, the goal shifts from agronomic traits to the precise manipulation of metabolic pathways to enhance the production of valuable Plant Natural Products (PNPs). Gene attenuation—the partial reduction of gene expression—is often more critical than complete gene knockout to avoid metabolic imbalances and maintain cell viability [43].
Pathway Engineering and Gene Attenuation
Secondary metabolites, such as alkaloids, flavonoids, and terpenoids, are crucial for human health but are often produced in low yields in plants [41] [39]. TALENs have been identified as a particularly powerful tool for this nuanced engineering, enabling targeted modifications to genes encoding key enzymes and transcription factors in these biosynthetic pathways [41]. For instance, TALEN-mediated genome editing has been successfully applied to modify pathways responsible for alkaloids and flavonoids in medicinal plants, unlocking new frontiers in secondary metabolite improvement [41].
While CRISPR is also widely used for metabolic engineering [39], TALENs are highlighted for their role in inducing genetic variation and facilitating hybridization in plant breeding programs aimed at enhancing the yield and quality of PNPs [41]. The ability to fine-tune gene expression through technologies like CRISPR interference (CRISPRi) allows for the optimization of metabolic flux without completely disrupting competing pathways, which is essential for maintaining the overall health of the plant cell factory [43].
The following diagram illustrates a generalized experimental workflow for conducting gene editing experiments in plants, from design to analysis.
Experimental Protocols and Methodologies
Robust experimental protocols are essential for successful gene editing outcomes. Below are detailed methodologies for key processes, drawing from standard practices in the field.
Protocol 1: Delivery of Editing Components into Plant Cells
Agrobacterium-mediated Transformation is a widely used method for delivering gene editing components into plant cells [42].
- Vector Construction: For CRISPR, the gene sequences for Cas9 and the gRNA(s) are cloned into a T-DNA binary vector under the control of plant-specific promoters. For TALENs or ZFNs, the sequences encoding the engineered proteins are cloned similarly [40].
- Agrobacterium Preparation: The constructed vector is introduced into Agrobacterium tumefaciens cells.
- Plant Material Co-cultivation: Explants (e.g., leaf discs, embryos) are harvested and immersed in the Agrobacterium suspension, allowing the bacteria to transfer the T-DNA containing the editing machinery into the plant cell genome.
- Selection and Regeneration: Explants are transferred to selection media containing antibiotics to eliminate non-transformed cells and hormones to promote callus formation and subsequent shoot regeneration.
- Rooting and Acclimatization: Developed shoots are transferred to rooting media, and resulting plantlets are acclimatized to greenhouse conditions.
Protocol 2: Analysis of Editing Efficiency and Specificity
- DNA Extraction: Genomic DNA is isolated from regenerated plantlets or tissue samples.
- PCR Amplification: The target genomic locus is amplified using specific primers.
- Sequencing Analysis:
- Sanger Sequencing: Followed by chromatogram decomposition software to identify indels.
- Next-Generation Sequencing (NGS): Provides a deep, quantitative measure of editing efficiency and can detect rare off-target events if the whole exome or genome is sequenced.
- Off-Target Assessment: Potential off-target sites are computationally predicted based on sequence similarity to the target site. These loci are then amplified and deep-sequenced to quantify any unintended mutations [19] [40].
The Scientist's Toolkit: Essential Research Reagents
Successful gene editing experiments rely on a suite of specialized reagents and tools. The following table details key solutions and their functions.
Table 3: Essential Research Reagents for Plant Genome Editing
| Research Reagent / Solution | Function / Application |
|---|---|
| T-DNA Binary Vectors | Plasmid vectors used in Agrobacterium-mediated transformation to deliver gene editing components (e.g., Cas9, gRNA, TALEN/ZFN genes) into plant cells [40]. |
| Plant Culture Media & Selection Agents | Nutrient media (e.g., Murashige and Skoog base) supplemented with plant growth regulators and antibiotics (e.g., kanamycin, hygromycin) for selecting transformed plant tissues [43]. |
| Restriction Enzymes & Cloning Kits | Molecular tools for assembling gene editing constructs, including Golden Gate assembly kits which are particularly useful for building TALEN arrays [40]. |
| PCR Kits & Sanger Sequencing Reagents | For amplifying and sequencing target loci to confirm successful genetic modifications and analyze editing efficiency. |
| Commercial Cell-Free Screening Kits | In vitro systems for testing the activity and specificity of designed nucleases (e.g., CRISPR gRNAs) before proceeding to plant transformation, saving time and resources. |
| Antibodies for Protein Detection | Western blot analysis using specific antibodies (e.g., anti-Cas9, anti-FLAG) to confirm the expression of editing proteins in transformed plant tissues [40]. |
| CRISPR-Cas12a/Cpf1 Systems | Alternative CRISPR systems with different PAM requirements, expanding the range of targetable sites in plant genomes [42]. |
| Base Editing & Prime Editing Systems | Advanced CRISPR-based tools that enable precise nucleotide changes without creating double-strand breaks, offering higher precision for metabolic engineering [44] [26]. |
ZFNs, TALENs, and CRISPR-Cas systems each occupy a unique niche in the plant genome editing landscape. CRISPR-Cas9 stands out for its unparalleled ease of use, cost-effectiveness, and suitability for high-throughput applications, making it the dominant platform for large-scale crop improvement programs aimed at enhancing yield, climate resilience, and nutritional content [38] [8]. TALENs offer robust performance and are particularly valued in applications requiring high specificity and for engineering complex traits, such as the modulation of secondary metabolite pathways in medicinal plants [41] [39]. ZFNs, as the pioneers, provided critical proof-of-concept but are now less frequently chosen for new projects due to their design complexity and cost [19] [8].
The future of plant genome editing lies in the continued refinement of these tools. For CRISPR, this includes the development of novel Cas variants with altered PAM requirements and reduced off-target effects, as well as the adoption of base and prime editors for ultimate precision [26] [8]. The integration of AI and big data into "Breeding 4.0" is set to further revolutionize the field, enabling predictive modeling of gene functions and optimal edit selection [42]. As regulatory frameworks evolve globally, these advanced technologies will play an increasingly vital role in building sustainable and resilient agricultural and pharmaceutical supply chains.
The transformative potential of therapeutic genome editing hinges on the efficient and safe delivery of editing machinery to target cells in vivo. While the scientific discourse often highlights the competitive advantages of clustered regularly interspaced short palindromic repeats (CRISPR) systems over zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) in terms of design simplicity and cost, their practical application is intrinsically linked to the delivery vector [2] [1] [45]. The choice between viral vectors and lipid nanoparticles (LNPs) is not merely a logistical decision; it profoundly influences the kinetics, distribution, and specificity of the gene editor, thereby directly impacting the safety and efficacy of the intervention. This guide provides a objective comparison of how LNP and viral vector delivery systems interact with the intrinsic specificity of ZFNs, TALENs, and CRISPR systems, synthesizing current data to inform preclinical development.
The fundamental challenge stems from the distinct molecular architectures of these editors. ZFNs and TALENs operate on a protein-DNA recognition principle, requiring the delivery of a nucleotide sequence encoding for a custom-designed protein for each new target [1] [9]. In contrast, CRISPR systems rely on RNA-DNA recognition, where a common Cas nuclease protein is directed by a easily programmable guide RNA (gRNA) [2] [45]. This core difference dictates their payload size and, consequently, their compatibility with different delivery vectors, which in turn affects their operational window and potential for off-target effects.
Before delving into delivery systems, it is crucial to understand the basic characteristics of the three primary editing platforms. The following table provides a consolidated comparison of their core mechanisms and performance metrics.
Table 1: Comparative Analysis of ZFNs, TALENs, and CRISPR-Cas9 Genome Editing Tools
| Parameter | ZFN | TALEN | CRISPR-Cas9 |
|---|---|---|---|
| DNA Recognition Mechanism | Protein-DNA [1] | Protein-DNA [1] | RNA-DNA [2] |
| Nuclease Component | FokI dimer [9] | FokI dimer [9] | Cas9 [2] |
| Target Site Length | 18-36 bp/ZFN pair [2] | 30-40 bp/TALEN pair [2] | ~22 bp [2] |
| Efficiency | Low to Moderate (0-12%) [2] | Moderate (0-76%) [2] | High (0-81%) [2] |
| Ease of Designing | Difficult [2] [9] | Difficult [2] [9] | Easy [2] [45] |
| Multiplexing Potential | Less Feasible [2] | Less Feasible [2] | Highly Feasible [2] |
| Affordability | Low [2] | Low [2] | High [2] |
| Reported Off-Target Effect | Less predictable [2] | Less predictable [2] | Highly predictable [2] |
| In Vivo Delivery System | AAV [2] | AAV [2] | AAV, Lentivirus [2] |
As illustrated, CRISPR-Cas9 offers significant advantages in design flexibility and cost. However, its reported off-target effects are a key point of consideration, the impact of which can be modulated by the choice of delivery vehicle.
Delivery Vector Platforms: Mechanisms and Trade-offs
The in vivo delivery of gene-editing components is primarily facilitated by two advanced platforms: Viral Vectors and Lipid Nanoparticles. Each system possesses unique characteristics that determine its suitability for different editors.
Viral Vectors: The Established Workhorse
Viral vectors, particularly adenoviruses (AdVs) and adeno-associated viruses (AAVs), are engineered to be replication-incompetent and used for their high transduction efficiency [46].
- Mechanism of Action: These vectors leverage the virus's natural ability to infect cells. The genetic material encoding the editor (e.g., ZFN, TALEN, or Cas9+gRNA) is packaged into the viral capsid. Upon systemic administration, the vector binds to specific cell surface receptors, enters the cell via endocytosis, and releases its genetic payload into the nucleus for transcription [46].
- Key Considerations: A primary safety concern with viral vectors is their potential to trigger adverse immunogenic responses, a risk tragically highlighted by a fatal immune response in a 1999 clinical trial [46]. Furthermore, the long-lasting expression of the editor, driven by viral promoters, can increase the risk of off-target effects and pathological insertional mutagenesis over time [46]. Their packaging capacity is also a major constraint; while AAV can typically carry ~4.7 kb, it is too small for certain TALEN pairs or for packaging SpCas9 with complex gRNA arrays, though smaller Cas orthologs like SaCas9 can be used [46] [45].
Lipid Nanoparticles (LNPs): The Non-Viral Rising Star
LNPs have emerged as a powerful non-viral alternative, proven clinically by their success in delivering siRNA (patisiran) and mRNA (COVID-19 vaccines) [46] [47].
- Mechanism of Action: LNPs are typically composed of four lipid components: an ionizable cationic lipid, PEG-lipid, phospholipid, and cholesterol [46]. The ionizable lipid is crucial for encapsulating the nucleic acid payload (e.g., mRNA for Cas9 and gRNA) through electrostatic interactions at low pH. Following systemic administration, LNPs accumulate at target sites, are internalized by cells via endocytosis, and the ionizable lipid facilitates endosomal escape, releasing the payload into the cytoplasm [46] [47].
- Key Considerations: A significant advantage of LNPs is their transient expression profile. mRNA delivered by LNPs is translated into protein but is not integrated into the genome and degrades naturally, limiting the window of editor activity and potentially reducing off-target effects [46]. They also exhibit low immunogenicity compared to viral vectors and have a much larger payload capacity, suitable for delivering the sizable CRISPR-Cas9 components [46]. However, achieving tissue-specific targeting beyond the liver remains an active area of research and development.
The diagram below summarizes the critical relationship between delivery vector properties and their impact on gene editor performance.
Experimental Data: Quantifying Delivery Impact on Specificity
The theoretical trade-offs between delivery systems are borne out in experimental data. The following table synthesizes quantitative findings from key studies that illuminate how vector choice interacts with editing outcomes.
Table 2: Experimental Data on Delivery Systems and Editing Outcomes
| Editing System | Delivery Vector | Experimental Model | Key Outcome Metrics | Reported Off-Target Findings |
|---|---|---|---|---|
| TALENs [48] | mRNA Electroporation | Human T Cells | TCR Knockout: Up to 81% of T cells [48] | Genome-wide analysis (IDLV capture) revealed only 3 off-target sites for the TALEN pair [48]. |
| CRISPR/Cas9 [48] | Plasmid Electroporation | Human T Cells | Not Specified | Genome-wide analysis (IDLV capture) revealed only 1 off-target site for one of the five gRNAs tested [48]. |
| CRISPR/Cas9 [46] [47] | LNP | Various in vivo models | Demonstrated successful in vivo editing; transient expression limits editing window. | The transient nature of LNP-delivered mRNA is hypothesized to reduce off-target effects by limiting nuclease exposure [46] [47]. |
| ZFN [9] | AAV | Human Pluripotent Stem Cells | Successful gene correction achieved. | A study identified one off-target mutation in 184 clones assessed, based on screening 10 potential sites [9]. |
Detailed Experimental Protocol: T Cell Receptor (TCR) Knockout
The high-specificity results for TALENs and CRISPR/Cas9 summarized in Table 2 were derived from a robust experimental workflow designed to engineer T cells for therapy [48].
- Objective: To knockout the endogenous αβ T cell receptor (TCR) in human T cells to prevent mispairing with transgenic TCRs introduced for cancer immunotherapy, thereby enhancing safety and efficacy.
- Nuclease Design and Preparation:
- Delivery and Transfection:
- Primary human T cells were isolated and activated.
- Delivery was achieved via electroporation for both TALEN mRNA and the CRISPR/Cas9 plasmid [48].
- Efficiency Assessment:
- Editing efficiency was quantified by flow cytometry to measure the loss of surface TCR expression (knockout) several days post-electroporation [48].
- Specificity Analysis (Genome-wide):
- The highly sensitive integrase-defective lentiviral vector (IDLV) capture method was used to empirically identify off-target sites across the genome in the treated T cells [48].
- This method involves transducing cells with IDLVs, which integrate preferentially into DSBs. The sites of integration are then sequenced to map all nuclease-induced breaks.
This protocol underscores that both TALENs and CRISPR/Cas9 can achieve high specificity, with the results being highly dependent on the specific target sequence and gRNA design.
The Scientist's Toolkit: Essential Reagents for Delivery and Editing Research
For researchers aiming to develop in vivo gene editing therapies, the following reagents and systems are critical components of the experimental toolkit.
Table 3: Essential Research Reagents for In Vivo Gene Editing Delivery
| Reagent / System | Function | Key Considerations |
|---|---|---|
| Ionizable Cationic Lipids (e.g., MC3) [46] | Core component of LNPs; encapsulates nucleic acid payload and enables endosomal escape. | Superior to permanently cationic lipids due to reduced toxicity and more efficient payload release. A key ingredient in the approved drug ONPATTRO [46]. |
| Adeno-Associated Virus (AAV) [2] [46] | Viral vector for in vivo delivery of editor genes. | Serotype determines tropism (e.g., AAV8 for liver). Limited packaging capacity (~4.7 kb) necessitates use of compact editors or split systems [46]. |
| In Vitro Transcription (IVT) Kits | Production of mRNA for Cas9 and gRNA for LNP delivery. | High-quality, capped, and polyadenylated mRNA is essential for high-yield translation and prolonged activity in vivo. |
| Homology-Directed Repair (HDR) Donor Template | Provides the correct sequence for precise gene correction after a DSB. | Can be delivered as a single-stranded oligodeoxynucleotide (ssODN) for small edits or as a double-stranded AAV vector for larger inserts [9]. |
| T7 Endonuclease I / NEXT Assay | Enzymatic method to detect nuclease-induced indels at predicted on- and off-target sites. | A rapid but indirect method for initial efficiency screening. Does not provide a comprehensive off-target profile [48]. |
| Integrase-Defective Lentiviral Vector (IDLV) Capture [48] | Genome-wide empirical method for identifying nuclease-induced DSBs. | Provides an unbiased, empirical map of off-target sites but is a complex cellular assay [48]. |
Integrated Discussion and Strategic Recommendations
The interplay between editor specificity and delivery vector is a cornerstone of therapeutic design. The data indicate that while CRISPR-Cas9 holds a dominant position due to its ease of use, both TALENs and ZFNs can achieve high specificity and efficiency, particularly in ex vivo settings like T cell engineering [48]. The choice of delivery system, however, can modulate the risk profile of even the most specific editor.
For instance, the persistent expression from AAV vectors, while beneficial for sustaining correction in dividing cells, presents a well-documented risk of increasing off-target mutations over time and eliciting immune responses against the viral capsid or the transgene [46]. Conversely, the transient delivery of CRISPR-Cas9 as mRNA via LNPs confines the editing activity to a short window. This transient action inherently limits the opportunity for off-target cleavage, making LNPs an attractive delivery modality for enhancing the in vivo specificity of CRISPR systems [46] [47]. This strategy, however, may require optimized conditions to achieve the desired level of on-target editing in the limited time frame.
Strategic Recommendations for Therapeutic Development
- For Large Payloads or High Transient Activity: LNP-mediated delivery of mRNA is the leading choice for CRISPR-Cas9 systems, offering a favorable safety profile and large payload capacity. This approach is particularly suitable for editing in non-dividing cells or for knock-out strategies where transient expression is sufficient.
- For Stable, Long-Term Expression in Post-Mitotic Tissues: AAV vectors remain a valuable tool, especially for delivering compact editors like ZFNs or smaller Cas orthologs (e.g., SaCas9) to tissues like the retina or central nervous system, where sustained expression is needed and the risk of immune response is mitigated.
- For Maximum Specificity with Protein-Based Editors: When the therapeutic objective demands the highest possible specificity and the target is amenable, electroporation of ZFN or TALEN mRNA ex vivo presents a robust and well-validated pathway, as demonstrated in T cell therapies.
In conclusion, the paradigm is shifting from a simple comparison of editing platforms to a holistic consideration of the editor-vector duo. The future of safe and effective in vivo genome editing lies in the rational matching of a high-specificity editor with a delivery vehicle whose pharmacokinetics and biodistribution are optimally tuned to the clinical application.
Navigating Challenges: Off-Target Effects and Strategies for Enhanced Precision
The advent of programmable nucleases has revolutionized genetic engineering, offering unprecedented control over genomic modifications. Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and CRISPR-Cas systems represent three generations of genome editing technologies, each with distinct mechanisms and specificities [49] [26]. While these tools have demonstrated remarkable potential across basic research and clinical applications, their safety profiles—particularly concerning off-target effects—vary significantly. Off-target effects refer to unintended genetic modifications at locations other than the intended target site, which can include small insertions/deletions (indels), point mutations, and more concerningly, large structural variations (SVs) including chromosomal translocations and megabase-scale deletions [50]. As gene editing technologies advance toward therapeutic applications, rigorous assessment and comparison of these off-target effects have become paramount for ensuring clinical safety [51].
The fundamental differences in how these editing platforms recognize DNA targets directly influence their off-target potential. ZFNs and TALENs rely on protein-DNA interactions, where customized protein domains recognize specific DNA sequences [49]. In contrast, CRISPR-Cas systems utilize RNA-DNA base pairing, where a guide RNA (gRNA) directs the Cas nuclease to complementary DNA sequences [49]. This distinction contributes to varying off-target profiles and necessitates specialized detection methodologies tailored to each platform's unique characteristics. This article provides a comprehensive comparison of current methodologies for quantifying off-target effects across these major editing platforms, with particular emphasis on their applications in therapeutic development.
Mechanism of Action and Off-target Profiles
Table 1: Comparison of Major Gene Editing Platforms
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| Target Recognition | Protein-DNA (zinc finger domains recognize DNA triplets) | Protein-DNA (TALE repeats recognize single nucleotides) | RNA-DNA (gRNA base pairing with DNA) |
| Nuclease Component | FokI dimer | FokI dimer | Cas9 single nuclease |
| Specificity Determinants | DNA triplet recognition by zinc fingers, requirement for dimerization | Single nucleotide recognition by TALE repeats, requirement for dimerization | gRNA complementarity, PAM sequence, chromatin accessibility |
| Primary Off-target Concerns | Off-target cleavage at similar sequences, cellular toxicity | Off-target cleavage at similar sequences | Mismatch tolerance in gRNA (especially distal end), non-canonical PAM recognition, DNA/RNA bulges |
| Ease of Design | Complex, limited by guanine-rich preference | Modular but repetitive assembly challenging | Simple, highly programmable |
| Therapeutic Validation | Clinical success in HIV (CCR5 disruption) | Preclinical and clinical studies | First FDA-approved therapies (e.g., Casgevy for SCD) |
ZFNs and TALENs operate through similar mechanisms, employing the FokI nuclease domain that requires dimerization for activity, which inherently increases specificity [26]. ZFNs recognize DNA via zinc finger proteins, each binding to a 3-base pair sequence, while TALENs utilize TALE repeats where each repeat recognizes a single nucleotide [26]. This protein-DNA interaction provides high specificity but makes redesign challenging and time-consuming. CRISPR-Cas9 systems, in contrast, achieve targeting through Watson-Crick base pairing between a guide RNA and DNA target sequence, significantly simplifying redesign but introducing different specificity challenges [49].
The specificity of CRISPR-Cas9 is influenced by multiple factors, including protospacer adjacent motif (PAM) recognition and the degree of complementarity between the gRNA and target DNA [49]. The seed sequence (PAM-proximal 10-12 nucleotides) is particularly critical for specific recognition and cleavage [49]. CRISPR-Cas9 can tolerate mismatches, especially in the distal region from the PAM, with evidence of cleavage even with up to six base mismatches [49]. Additional factors contributing to off-target activity include DNA/RNA bulges (extra nucleotide insertions from imperfect complementarity) and genetic diversity such as single nucleotide polymorphisms (SNPs) that may create novel off-target sites [49].
Beyond Small Indels: The Landscape of Unintended Edits
Traditional assessments of off-target effects have focused primarily on small indels at predicted off-target sites. However, advanced detection methods have revealed a more complex picture, including large structural variations (SVs), chromosomal translocations, megabase-scale deletions, and chromothripsis [50]. These unintended consequences pose substantial safety concerns for therapeutic applications, particularly when tumor suppressor genes or oncogenes are affected [50].
Notably, while off-target effects have received significant attention, on-target genomic aberrations deserve equal consideration. Large deletions at on-target sites can remove critical cis-regulatory elements with profound functional consequences [50]. Furthermore, strategies to enhance editing efficiency, such as using DNA-PKcs inhibitors to promote homology-directed repair (HDR), have been shown to exacerbate genomic aberrations, increasing both the frequency and scale of structural variations [50].
Methodologies for Detecting Off-Target Effects
Classification of Detection Approaches
Methodologies for identifying off-target effects can be broadly categorized into in silico (computational prediction), biochemical (in vitro), and cellular (in vivo) approaches [49] [52]. Each category offers distinct advantages and limitations, leading researchers to often employ complementary methods for comprehensive off-target assessment.
Table 2: Comparison of Major Off-target Detection Methodologies
| Method | Approach Category | Detection Principle | Strengths | Limitations |
|---|---|---|---|---|
| In silico Tools (Cas-OFFinder, CRISPOR) | Computational | Genome sequence analysis based on similarity, PAM rules, predictive models | Fast, inexpensive, useful for guide design | Predictions only; misses chromatin, repair, and nuclease activity influences |
| Digenome-seq | Biochemical (in vitro) | Whole-genome sequencing of Cas9-digested genomic DNA | Moderate sensitivity; uses purified genomic DNA | May overestimate cleavage; lacks biological context |
| CIRCLE-seq | Biochemical (in vitro) | Circularization and exonuclease enrichment of cleavage sites | High sensitivity; low input DNA requirement | Biochemical context only; may detect biologically irrelevant sites |
| GUIDE-seq | Cellular (in vivo) | Captures double-stranded oligodeoxynucleotide integration at DSB sites | High biological relevance; captures cellular context | Requires efficient delivery; may miss rare off-target sites |
| DISCOVER-seq | Cellular (in vivo) | Identifies MRE11 recruitment to DSB sites via ChIP-seq | Captures real-time breaks in native chromatin | Moderate sensitivity; complex workflow |
| BLESS | Cellular (in situ) | In situ labeling of DSB ends with biotin linkers | Preserves genomic architecture; captures breaks in situ | Technically challenging; lower throughput |
Experimental Protocols for Key Detection Methods
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) is a highly sensitive cellular method that captures off-target effects in living cells [52] [53]. The protocol involves: (1) Transfection of cells with CRISPR-Cas9 components along with a double-stranded oligodeoxynucleotide (dsODN) tag; (2) Integration of the dsODN into double-strand break (DSB) sites during repair; (3) Genomic DNA extraction and fragmentation; (4) Enrichment and amplification of tagged fragments; (5) Next-generation sequencing and computational analysis to identify off-target integration sites [53]. GUIDE-seq provides comprehensive genome-wide DSB mapping but requires efficient delivery of both editing components and the dsODN tag into cells.
CIRCLE-seq (Circularization for In vitro Reporting of Cleavage Effects by Sequencing) is an ultra-sensitive biochemical approach conducted in vitro [52]. The methodology includes: (1) Extraction and purification of genomic DNA; (2) Circularization of DNA fragments using single-stranded DNA ligase; (3) In vitro digestion with Cas9-sgRNA ribonucleoprotein (RNP) complexes; (4) Exonuclease treatment to degrade linear DNA while preserving cleaved circular fragments; (5) Library preparation and high-throughput sequencing of enriched cleavage products [52]. CIRCLE-seq offers exceptional sensitivity for detecting rare off-target sites but may identify cleavages that do not occur in cellular contexts due to the absence of chromatin structure and DNA repair mechanisms.
DISCOVER-seq (Discovery of In Situ Cleavage Sites by Sequencing) leverages endogenous DNA repair machinery to identify off-target effects in native chromatin contexts [52]. The protocol involves: (1) Editing of target cells with CRISPR-Cas9; (2) Cross-linking and chromatin immunoprecipitation (ChIP) using antibodies against MRE11, a key protein recruited to DSB sites; (3) Immunoprecipitated DNA purification and sequencing; (4) Bioinformatic analysis to identify MRE11-enriched genomic regions corresponding to Cas9 cleavage sites [52]. This method captures off-target activity under physiological conditions but may have limited sensitivity for detecting transient or rapidly repaired breaks.
(Classification of Major Off-target Detection Methodologies)
Quantitative Comparison of Detection Method Performance
Table 3: Sensitivity and Practical Considerations of Off-target Assays
| Method | Detection Sensitivity | Input DNA Requirement | Throughput | Biological Relevance | Key Applications |
|---|---|---|---|---|---|
| In silico Prediction | N/A (computational) | N/A | High | Low | Initial sgRNA design and risk assessment |
| Digenome-seq | Moderate (requires deep sequencing) | Micrograms of genomic DNA | Moderate | Low | Genome-wide in vitro cleavage mapping |
| CIRCLE-seq | High (detects rare off-targets) | Nanograms of genomic DNA | High | Low | Comprehensive in vitro off-target discovery |
| CHANGE-seq | Very high (reduced false negatives) | Nanograms of genomic DNA | High | Low | Sensitive in vitro profiling with tagmentation |
| GUIDE-seq | High for cellular context | Cellular DNA from edited cells | Moderate | High | Biologically relevant off-target identification |
| DISCOVER-seq | Moderate | Cellular DNA; ChIP-seq | Moderate | High | Real-time break mapping in native chromatin |
| BLESS | Moderate (limited by labeling efficiency) | Fixed cells/permeabilized nuclei | Low | Medium | Spatial mapping of breaks in genomic architecture |
Sensitivity varies considerably across detection methods, with biochemical approaches generally offering higher theoretical sensitivity but potentially overestimating biologically relevant off-target activity [52]. Cellular methods, while capturing the influence of chromatin structure and DNA repair pathways, may miss rare off-target events due to limited sampling depth or delivery efficiency [52]. The limit of detection for methods like CIRCLE-seq and CHANGE-seq can reach sub-nanogram DNA inputs, enabling identification of extremely rare cleavage events [52].
Recent advances have highlighted the importance of unbiased genome-wide approaches over biased methods that only examine predicted off-target sites [52]. This is particularly relevant for clinical applications, as demonstrated during the FDA review of exa-cel (Casgevy), where concerns were raised about whether in silico prediction databases adequately represented genetic diversity across patient populations [52]. Regulatory agencies now recommend multiple complementary methods for comprehensive off-target assessment in therapeutic development [52].
Advanced Considerations in Off-target Assessment
Structural Variations and Complex Rearrangements
Beyond simple indels, CRISPR-Cas9 editing can induce large structural variations (SVs) including chromosomal translocations, megabase-scale deletions, and chromothripsis [50]. These complex rearrangements pose significant safety concerns but are often undetected by conventional amplicon sequencing approaches that rely on short-read technologies [50]. Methods such as CAST-Seq and LAM-HTGTS have been developed specifically to identify these larger-scale genomic alterations [50].
The use of DNA-PKcs inhibitors to enhance HDR efficiency has been shown to dramatically increase the frequency of these structural variations. One study reported a thousand-fold increase in chromosomal translocations with AZD7648 treatment, highlighting the potential risks of manipulating DNA repair pathways to improve editing outcomes [50]. Traditional quantification methods may overestimate HDR efficiency when large deletions remove primer binding sites, rendering these aberrations invisible to standard PCR-based assays [50].
Platform-Specific Off-target Mitigation Strategies
Each editing platform has inspired specific strategies to minimize off-target effects:
For CRISPR-Cas9: (1) High-fidelity Cas9 variants (e.g., SpCas9-HF1, eSpCas9, HypaCas9) with reduced non-specific DNA binding; (2) Truncated sgRNAs with shorter complementarity regions; (3) Cas9 nickase approaches requiring paired nicking for DSB formation; (4) dCas9-FokI fusions that leverage FokI dimerization requirements [49] [50]. Additionally, base editing and prime editing technologies offer alternative approaches that can minimize DSB formation altogether [26].
For ZFNs and TALENs: Optimization primarily focuses on enhancing DNA-binding specificity through improved domain design and selection. The inherent requirement for dimerization of FokI nuclease domains provides a natural specificity checkpoint not present in standard CRISPR-Cas9 systems [26].
(DNA Repair Pathways and Editing Outcomes)
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Off-target Assessment
| Reagent/Category | Specific Examples | Function in Off-target Analysis |
|---|---|---|
| Cas9 Nuclease Variants | Wild-type SpCas9, SpCas9-HF1, eSpCas9, HiFi Cas9 | Engineered for enhanced specificity; reduce off-target cleavage while maintaining on-target activity |
| Specialized Detection Kits | GUIDE-seq kit, CIRCLE-seq kit | Provide optimized reagents and protocols for specific off-target detection methods |
| Library Prep Systems | Illumina Nextera, Swift Accel | Enable efficient NGS library construction from low-input DNA for sensitive off-target detection |
| Bioinformatics Tools | CRISPOR, Cas-OFFinder, CRISPResso2 | Predict potential off-target sites and analyze sequencing data to quantify editing outcomes |
| DNA Repair Modulators | DNA-PKcs inhibitors, 53BP1 inhibitors | Manipulate DNA repair pathways to study their influence on editing outcomes and off-target profiles |
| Control gRNAs/sgRNAs | Validated high-specificity and low-specificity guides | Serve as benchmarks for comparing off-target profiles of novel gRNA designs |
The selection of appropriate reagents is critical for robust off-target assessment. High-fidelity Cas9 variants such as HiFi Cas9 have demonstrated significantly reduced off-target activity while maintaining robust on-target editing [50]. For detection methods, specialized library preparation kits optimized for low-input DNA can enhance sensitivity and reduce bias in methods like CIRCLE-seq and GUIDE-seq [52].
Bioinformatics tools constitute an essential component of the off-target assessment toolkit. CRISPOR utilizes cutting frequency determination (CFD) scores incorporating position-specific mismatch tolerance weights to predict off-target sites [53]. More recently, machine learning approaches like CRISOT have attempted to improve prediction accuracy by modeling intermolecular and intramolecular interactions during Cas9 binding, though performance challenges remain with PR-AUC values typically between 0.3-0.5 [53].
Comprehensive assessment of off-target effects remains a critical challenge in therapeutic genome editing. While each major editing platform—ZFNs, TALENs, and CRISPR-Cas systems—presents distinct off-target profiles, methodological advances have significantly enhanced our ability to quantify these unintended effects. Biochemical methods like CIRCLE-seq offer exceptional sensitivity for in vitro off-target discovery, while cellular methods such as GUIDE-seq and DISCOVER-seq provide crucial insights into biologically relevant editing outcomes in native chromatin contexts.
The field continues to evolve toward more comprehensive assessment strategies that detect not only small indels but also large structural variations through methods like CAST-Seq and LAM-HTGTS [50]. As regulatory frameworks mature, standardized off-target assessment employing complementary methodologies will be essential for advancing gene editing therapies. The recent FDA approval of the first CRISPR-based therapy marks a significant milestone while highlighting the ongoing importance of rigorous safety evaluation [52]. Future directions will likely focus on developing even more sensitive detection methods, standardizing assessment protocols across platforms, and improving computational prediction tools through advanced machine learning approaches that incorporate both sequence features and cellular context.
The advent of targeted nucleases has revolutionized genetic engineering, enabling precise modifications of DNA sequences for research and therapeutic applications. Among these technologies, Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs) represent the first generation of programmable nucleases, while CRISPR-Cas9 has emerged as a more recent and transformative platform [8]. Each system operates through distinct mechanisms to create double-strand breaks (DSBs) in DNA, but they differ significantly in their design, implementation, and associated challenges.
ZFNs and TALENs both rely on protein-DNA interactions for target recognition. ZFNs utilize zinc finger domains, each recognizing approximately three nucleotide triplets, fused to the FokI nuclease domain. TALENs employ transcription activator-like effector (TALE) repeats, where each repeat recognizes a single nucleotide, providing greater design flexibility than ZFNs [8]. Both systems require complex protein engineering for each new target sequence. In contrast, the CRISPR-Cas9 system utilizes a guide RNA (gRNA) that pairs with complementary DNA sequences to direct the Cas9 nuclease to specific genomic loci [54]. This RNA-based targeting mechanism simplifies design and implementation but introduces unique challenges, particularly regarding off-target effects and immune recognition.
The specificity of gene editing technologies is paramount for their therapeutic application. While all nucleases can potentially cleave off-target sites, the mechanisms and frequencies differ substantially. This article provides a comprehensive comparison of CRISPR-specific challenges—gRNA-dependent off-targeting and immune responses to Cas9—contrasting them with the limitations of ZFNs and TALENs to provide researchers with a clear framework for technology selection.
gRNA-Dependent Off-Target Effects: Mechanisms and Comparisons
Fundamental Mechanisms of Off-Target Effects
CRISPR-Cas9's off-target effects stem primarily from the flexibility of gRNA-DNA interactions. The gRNA can tolerate mismatches, particularly in the distal 5' region, bulges, and gaps, while still facilitating Cas9 binding and cleavage [51]. This permissiveness can lead to cleavage at sites with significant sequence similarity to the intended target. The frequency and severity of these off-target events are influenced by multiple factors, including gRNA sequence composition, chromatin accessibility, Cas9 expression levels, and delivery method [54].
The fundamental difference between CRISPR and traditional nucleases lies in their recognition mechanisms. ZFNs and TALENs achieve specificity through protein-DNA interactions, which typically require more exact matching. The FokI nuclease domains in both ZFNs and TALENs must dimerize to become active, adding an additional layer of specificity as two independent DNA binding events must occur in close proximity [8]. While this doesn't eliminate off-target effects entirely, it constrains their potential locations.
Table 1: Comparison of Off-Target Mechanisms Across Gene Editing Platforms
| Feature | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Targeting Mechanism | RNA-DNA base pairing | Protein-DNA recognition | Protein-DNA recognition |
| Specificity Determinants | gRNA complementarity, PAM sequence | TALE repeat recognition, FokI dimerization | Zinc finger recognition, FokI dimerization |
| Mismatch Tolerance | High (especially in 5' gRNA region) | Low | Low |
| Primary Off-Target Concerns | Sites with similar sequences to gRNA | Homodimerization at unrelated sites | Cross-reactivity of zinc fingers |
| Influence of Chromatin State | High | Moderate | Moderate |
Quantitative Comparisons of Editing Specificity
Direct comparisons between editing platforms reveal distinct specificity profiles. CRISPR-Cas9 generally exhibits higher editing efficiency but often at the cost of increased off-target activity compared to TALENs. In a study targeting the CCR5 gene, TALENs demonstrated high specificity with minimal off-target effects, while CRISPR showed greater efficiency but required comprehensive off-target analysis [8].
Beyond simple mismatches, CRISPR systems can induce more severe genomic alterations. Recent studies have revealed that CRISPR editing can generate large structural variations (SVs), including kilobase- to megabase-scale deletions, chromosomal translocations, and chromothripsis [50]. These aberrations occur not only at on-target sites but also at off-target locations, raising substantial safety concerns for therapeutic applications. While similar effects have been observed with ZFNs and TALENs, they have been more extensively documented with CRISPR systems due to their widespread adoption and comprehensive analysis [50].
The detection of these off-target events presents methodological challenges. Traditional short-read sequencing approaches often fail to identify large deletions and rearrangements that eliminate primer binding sites, leading to underestimation of off-target frequencies [50]. Advanced methods like CAST-Seq and LAM-HTGTS have been developed to better characterize these structural variations, providing more comprehensive safety profiles [50].
Immune Responses to Cas9: A Unique Challenge for CRISPR Therapies
Origins and Prevalence of Anti-Cas9 Immunity
The immunogenicity of Cas9 proteins represents a distinctive challenge for CRISPR-based therapies not shared by ZFNs or TALENs. As bacterial-derived proteins, Cas9 nucleases are recognized as foreign by the human immune system, potentially triggering both innate and adaptive immune responses [55]. Approximately 80% of people have pre-existing immunity to Cas9 proteins from common bacterial exposures, particularly to Cas9 from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) [56].
This pre-existing immunity poses multiple risks for therapeutic applications. Immune recognition can lead to rapid clearance of Cas9-expressing cells, reducing editing efficiency. It may also trigger inflammatory responses and serious adverse effects, particularly for in vivo applications where the editing components are delivered directly into the body [55]. A Grade 4 liver toxicity event reported in a Phase 3 trial of a CRISPR-Cas therapy for transthyretin amyloidosis highlights the potential clinical significance of these immune responses, though the exact role of immunity in this case remains under investigation [57].
Comparative Immunogenicity Profiles
Unlike CRISPR-Cas9, ZFNs and TALENs are engineered from human transcription factor domains fused to the FokI nuclease. Since these components are largely derived from eukaryotic sources, they exhibit lower immunogenicity than the bacterial-derived Cas9 [11]. This fundamental difference in origin translates to distinct immune recognition profiles that significantly impact therapeutic development.
Table 2: Immune Response Comparison Across Gene Editing Platforms
| Immune Parameter | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Origin of DNA-Binding Domain | Bacterial | Bacterial/Plant (Xanthomonas) | Eukaryotic (Human) |
| Pre-existing Immunity in Human Population | High (~80%) | Low | Very Low |
| Immune Recognition Elements | Multiple immunogenic epitopes identified | Limited data | Minimal epitopes |
| Impact on Therapeutic Efficacy | Potential reduction due to immune clearance | Minimal evidence of impact | Minimal evidence of impact |
| Risk of Inflammatory Responses | Significant for in vivo applications | Low | Low |
Experimental Evidence and Clinical Correlations
Research led by Feng Zhang's team at the Broad Institute has systematically identified the molecular basis of Cas9 immunogenicity. Using mass spectrometry, they pinpointed specific immunogenic sequences in both SpCas9 and SaCas9—short peptides approximately eight amino acids long that trigger immune recognition [56]. This detailed mapping of epitopes represents a crucial step toward engineering less immunogenic Cas9 variants.
Experimental studies in humanized mouse models have demonstrated that immune responses can substantially reduce the efficacy of CRISPR therapies. In contrast, similar assessments of ZFNs and TALENs have not revealed comparable immune-mediated clearance, aligning with their more human-like protein sequences [11]. This immunological challenge has prompted the development of creative solutions, including engineered Cas variants with reduced immunogenicity and transient delivery methods that limit exposure to the immune system.
Mitigation Strategies and Experimental Approaches
Computational Design and Protein Engineering
Multiple strategies have emerged to address CRISPR's unique challenges. For off-target effects, computational gRNA design tools have become increasingly sophisticated, incorporating mismatch penalties, chromatin accessibility data, and epigenetic markers to predict and avoid problematic target sites [51]. Additionally, high-fidelity Cas9 variants (e.g., HiFi Cas9) with reduced off-target activity have been engineered through rational design [50].
To address immunogenicity, researchers have developed epitope-engineered Cas9 versions that evade immune detection while maintaining editing function. Using structure-based computational tools, scientists have designed Cas9 and Cas12 proteins with modified surface residues in immunogenic regions, resulting in significantly reduced immune responses in humanized mouse models [56]. These engineered nucleases represent a promising approach for safer in vivo applications.
Alternative CRISPR systems also offer potential solutions. Base editing and prime editing technologies can introduce precise changes without creating double-strand breaks, significantly reducing off-target effects [44]. In a murine model of sickle cell disease, base editing demonstrated higher editing efficiency and fewer genotoxicity concerns compared to standard CRISPR-Cas9 [57]. Similarly, compact Cas variants like Cas12f have been engineered for improved specificity while maintaining small size for delivery applications [57].
Experimental Protocols for Assessing Specificity and Immunogenicity
Off-Target Assessment Using CAST-Seq
Principle: The CAST-Seq (Circularization for Amplification and Sequencing of Off-Target Sites) method comprehensively identifies CRISPR-induced structural variations and translocations [50].
Procedure:
- Cell Processing: Treat cells with CRISPR-Cas9 components (e.g., via electroporation)
- DNA Extraction: Harvest genomic DNA 72 hours post-treatment
- Restriction Digestion: Use four-cutter restriction enzymes for fragmentation
- Adapter Ligation: Add biotinylated adapters to fragment ends
- Circularization: Perform intramolecular ligation to create DNA circles
- Inverse PCR: Amplify rearranged junctions using target-specific primers
- Library Preparation and Sequencing: Prepare libraries for high-throughput sequencing
- Bioinformatic Analysis: Map chimeric reads to identify translocation partners and structural variations
This protocol enables sensitive detection of chromosomal rearrangements between on-target sites, off-target sites, and other genomic regions, providing a comprehensive safety profile [50].
Immune Recognition Assay Using Mass Spectrometry
Principle: Identify specific Cas9 epitopes recognized by human immune cells to guide protein engineering [56].
Procedure:
- Antigen Presentation: Incubate human dendritic cells with Cas9 protein
- T Cell Co-culture: Co-culture primed dendritic cells with autologous T cells
- MHC Complex Isolation: Immunoprecipitate MHC-peptide complexes
- Peptide Elution: Acid-elute bound peptides from MHC complexes
- Liquid Chromatography-Mass Spectrometry: Separate and analyze eluted peptides
- Epitope Mapping: Identify Cas9-derived peptides and their source sequences
- Validation: Synthesize candidate epitopes and test T cell activation
This approach enables precise mapping of immunogenic regions within Cas9, facilitating rational design of less immunogenic variants [56].
Research Reagent Solutions
Table 3: Essential Reagents for CRISPR Specificity and Immunogenicity Research
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| High-Fidelity Cas Variants | HiFi Cas9, HypaCas9 | Reduce off-target editing while maintaining on-target efficiency |
| Immuno-Evasive Cas9 | eCas9-IR, Deimmunized Cas9 | Engineered variants with reduced immunogenicity for in vivo applications |
| Off-Target Detection Kits | CAST-Seq kit, GUIDE-seq | Comprehensive identification of off-target sites and structural variations |
| Delivery Systems | LNP formulations, AAV variants | Efficient delivery while modulating immune responses |
| Immune Assessment Tools | MHC-peptide arrays, ELISpot kits | Evaluate T-cell responses to Cas9 and editing components |
| Control gRNAs | Validated high/low specificity gRNAs | Benchmark off-target profiles and assay performance |
| Bioinformatic Tools | Cas-OFFinder, CrispRGold | Predict potential off-target sites during gRNA design |
CRISPR-Cas9 has democratized gene editing with its simplicity and versatility, but its unique challenges—gRNA-dependent off-target effects and immune responses to bacterial Cas proteins—require careful consideration in research and therapeutic contexts. While ZFNs and TALENs offer alternative platforms with potentially superior specificity in certain applications and lack the pronounced immunogenicity of CRISPR, they come with their own limitations regarding design complexity and scalability.
The choice between these platforms depends heavily on the specific application. For large-scale functional screens, CRISPR's scalability and ease of design make it ideal. For clinical applications requiring minimal off-target risk, particularly in immunogenic contexts, TALENs or immuno-engineered CRISPR variants may be preferable. Emerging approaches like base editing and prime editing offer promising alternatives that circumvent both traditional off-target effects and immune activation.
As the field advances, the integration of machine learning for gRNA design, continued engineering of CRISPR components, and comprehensive safety assessment protocols will further enhance the specificity and safety of all gene editing platforms. Researchers should maintain a nuanced perspective on the relative advantages of each system, selecting the most appropriate tool based on their specific experimental or therapeutic goals while implementing robust controls to validate editing specificity and monitor immune responses.
The evolution of programmable genome editing tools has transformed biological research and therapeutic development. Beginning with Zinc-Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), and accelerating with the discovery of the CRISPR-Cas9 system, each technology platform has offered distinct advantages and limitations regarding specificity, efficiency, and ease of use [58]. While early CRISPR-Cas9 systems demonstrated remarkable programmability, concerns emerged regarding off-target effects—unintended modifications at genomic sites with sequences similar to the target site [59]. This comparative guide examines engineering solutions developed to address specificity challenges: high-fidelity Cas variants, base editors, and prime editors. Framed within the broader context of specificity comparisons between ZFNs, TALENs, and CRISPR systems, this analysis provides researchers with objective performance data and methodologies to inform experimental design and therapeutic development.
Comparative Specificity of Major Editing Platforms
Mechanisms of Target Recognition and Cleavage
The three major generations of genome editing tools—ZFNs, TALENs, and CRISPR-Cas systems—employ fundamentally different mechanisms for DNA recognition and cleavage, which directly impact their specificity profiles [58]:
- ZFNs utilize zinc-finger proteins, where each domain recognizes a 3-base pair DNA triplet. The FokI nuclease domain must dimerize to create a double-strand break (DSB), requiring two ZFN proteins to bind opposite DNA strands in close proximity [10].
- TALENs employ transcription activator-like effector (TALE) repeats, where each repeat recognizes a single nucleotide. Like ZFNs, TALENs use the FokI nuclease that requires dimerization for DSB formation [10].
- CRISPR-Cas9 relies on RNA-DNA complementarity through a guide RNA (gRNA) that directs the Cas nuclease to target sequences adjacent to a Protospacer Adjacent Motif (PAM) [58].
Experimental Comparison of Off-Target Activities
A direct comparative study using Genome-wide Unbiased Identification of DSBs Enabled by Sequencing (GUIDE-seq) to evaluate off-target activities of ZFNs, TALENs, and SpCas9 at the human papillomavirus 16 (HPV16) genome revealed significant differences in specificity [60].
Table 1: Off-Target Counts Detected by GUIDE-seq in HPV16 Genes
| Editing Platform | URR Gene | E6 Gene | E7 Gene |
|---|---|---|---|
| SpCas9 | 0 | 0 | 4 |
| TALENs | 1 | 7 | 36 |
| ZFNs | 287 | - | - |
The study demonstrated that SpCas9 generally exhibited higher specificity than ZFNs and TALENs, with ZFNs showing substantial off-target activity (287 off-target sites in the URR gene) [60]. The variability in ZFN specificity was correlated with the count of middle "G" in zinc finger proteins, while TALEN designs that improved efficiency (e.g., αN or NN) inevitably increased off-target effects [60].
High-Fidelity Cas Variants
Engineering Strategies for Enhanced Specificity
High-fidelity Cas variants were developed to reduce off-target effects while maintaining on-target efficiency through protein engineering approaches [61]. These engineered Cas9 variants recognize the same PAM sequences as wild-type SpCas9 but exhibit reduced off-target activity through various molecular strategies [58]:
- Reduced non-specific DNA binding: Weakening the interaction between Cas9 and the DNA backbone to decrease binding at non-target sites.
- Enhanced conformational proofreading: Requiring more perfect complementarity between the gRNA and target DNA for activation.
- Dimeric nickase systems: Using paired Cas9 nickases that create single-strand breaks only when both gRNAs bind adjacent sites.
Table 2: High-Fidelity Cas9 Variants and Their Specificity Profiles
| Variant | Key Mutations | Specificity Improvement | On-Target Efficiency |
|---|---|---|---|
| eSpCas9 | K848A, K1003A, R1060A | 10- to 100-fold reduction in off-targets | Comparable to wild-type |
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | >85% reduction in off-target activity | Slightly reduced in some contexts |
| HypaCas9 | N692A, M694A, Q695A, H698A | Enhanced proofreading mechanism | Maintained high efficiency |
| evoCas9 | 32 mutations from directed evolution | 100-fold higher fidelity | 70-95% of wild-type activity |
Experimental Validation of High-Fidelity Variants
Methodology for Off-Target Assessment: The GUIDE-seq protocol provides a robust methodology for unbiased genome-wide off-target detection [60]. The experimental workflow involves:
- Transfection of cells with nuclease components and a double-stranded oligodeoxynucleotide (dsODN) tag
- Capture and integration of dsODN tags at nuclease-induced double-strand break sites
- PCR amplification and next-generation sequencing of tagged sites
- Bioinformatics analysis to identify off-target sites
Key Findings: Studies implementing these validation methods have demonstrated that high-fidelity Cas variants can significantly reduce off-target effects while maintaining therapeutic efficacy. For example, HypaCas9 showed substantially improved specificity while maintaining robust on-target activity across multiple genomic loci [58]. The reduced off-target effects of these engineered variants make them particularly valuable for therapeutic applications where precision is critical.
Base Editing Systems
Mechanism and Design Principles
Base editors represent a distinct approach to precision genome editing that avoids double-strand breaks entirely. These systems utilize catalytically impaired Cas proteins (dCas9 or Cas9 nickase) fused to nucleobase deaminase enzymes that enable direct chemical conversion of one DNA base to another [61] [62].
The two primary classes of DNA base editors are:
- Cytosine Base Editors (CBEs): Convert cytosine to thymine (C•G to T•A)
- Adenine Base Editors (ABEs): Convert adenine to guanine (A•T to G•C)
Base editors combine targeting components with cellular repair mechanisms to achieve precise nucleotide conversions without double-strand breaks. CBEs utilize cytidine deaminase enzymes coupled with uracil glycosylase inhibitor (UGI) to prevent repair of the edited base, while ABEs use engineered adenine deaminases to achieve A•T to G•C conversions [61] [62].
Specificity Advantages and Limitations
Base editing offers significant specificity advantages over conventional CRISPR-Cas9 nuclease approaches. By avoiding double-strand breaks, base editors eliminate the formation of indels and large genomic rearrangements associated with DSB repair [59]. Studies using genome-wide off-target analysis by two-cell embryo injection (GOTI) have shown that advanced base editors like AccuBase can achieve near-zero off-target effects, with single-nucleotide variant (SNV) counts similar to negative controls [59].
However, base editors have certain limitations:
- Restricted to four transition mutations (C→T, T→C, A→G, G→A) without capability for transversions, insertions, or deletions [62]
- Potential for bystander edits where nearby editable bases within the activity window are modified
- Off-target effects can still occur in RNA or through certain DNA deaminase-independent mechanisms
Prime Editing Systems
Mechanism and Molecular Components
Prime editing represents a versatile "search-and-replace" genome editing technology that can install all 12 possible base-to-base conversions, small insertions, and small deletions without requiring double-strand breaks or donor DNA templates [62] [63]. The system consists of three key components:
- Prime Editing Guide RNA (pegRNA): Specifies the target site and encodes the desired edit
- Cas9 Nickase (H840A): Creates a single-strand nick in the DNA
- Reverse Transcriptase: Synthesizes DNA using the pegRNA as a template
The prime editing process involves: (1) binding of the prime editor complex to the target DNA and nicking of the non-target strand, (2) hybridization of the primer binding site (PBS) to the nicked DNA, (3) reverse transcription using the template embedded in the pegRNA, and (4) resolution and repair of the DNA heteroduplex to permanently incorporate the edit [62] [63].
Specificity Profile and Validation
Prime editing demonstrates exceptional specificity with significantly reduced off-target effects compared to conventional CRISPR-Cas9 systems. This enhanced precision stems from multiple molecular features:
- Avoidance of double-strand breaks and associated error-prone repair pathways
- Requirement for three separate hybridization events (target recognition, primer binding, and template alignment) for successful editing
- Reduced cellular toxicity and DNA damage response
In a landmark study correcting the sickle cell disease mutation, prime editing achieved precise correction in 40% of patient-derived stem cells without detectable off-target effects [62]. The technology has shown particular promise for therapeutic applications requiring high precision, such as correcting point mutations associated with genetic disorders.
Comparative Performance Analysis
Quantitative Comparison of Editing Platforms
Table 3: Comprehensive Comparison of Genome Editing Platforms and Their Specificity Profiles
| Editing Platform | Editing Type | DSB Formation | Off-Target Rate | Therapeutic Applications | Key Limitations |
|---|---|---|---|---|---|
| ZFNs | DSB-dependent | Yes | High (287 off-targets in URR) [60] | HIV (CCR5 disruption) [60] | Complex design, high toxicity, limited targeting |
| TALENs | DSB-dependent | Yes | Medium (1-36 off-targets) [60] | B-ALL (UCART19) [60] | Large size, challenging delivery |
| CRISPR-Cas9 | DSB-dependent | Yes | Variable | Sickle cell disease, beta-thalassemia [8] | gRNA-dependent off-target effects |
| High-Fidelity Cas Variants | DSB-dependent | Yes | 10-100x reduction vs wild-type [58] | Requiring high precision | Potential reduced on-target efficiency |
| Base Editors | Single-base conversion | No | Near-zero (advanced editors) [59] | Sickle cell disease (BEAM-101) [59] | Limited to transition mutations, bystander edits |
| Prime Editors | Search-and-replace | No | Lowest reported [62] | Proof-of-concept for various mutations [63] | Lower efficiency, complex pegRNA design |
Applications in Therapeutic Contexts
The choice of editing platform depends heavily on the specific therapeutic application and precision requirements:
- For gene disruption (e.g., CCR5 for HIV resistance): High-fidelity Cas variants provide an optimal balance of efficiency and specificity [60] [58]
- For point mutation correction (e.g., sickle cell disease): Base editors offer efficient correction with minimal indel formation [62] [59]
- For complex edits requiring precision (e.g., multiple base changes or small insertions): Prime editors provide the greatest versatility without DSBs [63]
Recent advances have demonstrated the potential of combining these approaches, such as multiplex base editing strategies that simultaneously target two BCL11A enhancers to treat sickle cell disease, achieving superior fetal hemoglobin reactivation while avoiding genomic rearrangements associated with traditional CRISPR-Cas9 nucleases [64].
Research Reagent Solutions
Essential Reagents for Genome Editing Studies
Table 4: Key Research Reagents for High-Specificity Genome Editing
| Reagent Category | Specific Examples | Function | Considerations for Specificity |
|---|---|---|---|
| Nuclease Enzymes | HiFi Cas9, HypaCas9, BE4max, PE2 | Core editing machinery | High-fidelity variants reduce off-target effects |
| Guide RNAs | sgRNAs, pegRNAs | Target specification | Modified scaffolds enhance specificity [61] |
| Delivery Vehicles | AAVs, LNPs, Electroporation | Intracellular delivery | Transient delivery reduces off-target exposure [61] |
| Off-Target Detection | GUIDE-seq, GOTI, CIRCLE-seq | Specificity validation | Unbiased genome-wide profiling essential |
| Cell Lines | HEK293, iPSCs, Primary T cells | Experimental models | Primary cells more relevant but harder to edit [12] |
Experimental Design for Specificity Assessment
Robust assessment of editing specificity requires implementation of rigorous experimental protocols:
GUIDE-seq Protocol [60]:
- Transfect cells with nuclease components and dsODN tag
- Allow 72-96 hours for tag integration at DSB sites
- Extract genomic DNA and prepare sequencing library
- Sequence and analyze using bioinformatics pipelines
- Validate potential off-target sites by targeted sequencing
Key Considerations:
- Include both positive and negative controls
- Use multiple computational prediction tools alongside empirical methods
- Assess off-target effects in biologically relevant cell types
- Consider time-dependent effects on off-target detection
The landscape of genome editing technologies has evolved significantly toward enhanced specificity without sacrificing functionality. While standard CRISPR-Cas9 systems already demonstrate favorable specificity profiles compared to earlier technologies like ZFNs and TALENs, the development of high-fidelity Cas variants, base editors, and prime editors has dramatically expanded the toolbox for precision genome engineering. Each platform offers distinct advantages for particular applications—from efficient gene disruption with high-fidelity nucleases to precise single-base conversion with base editors and versatile "search-and-replace" capabilities with prime editors. As these technologies continue to mature and undergo rigorous safety profiling, they hold tremendous promise for therapeutic applications requiring exceptional precision, ultimately enabling new treatments for genetic disorders with improved safety profiles.
The Role of AI and Machine Learning in Predicting gRNA Efficiency and Off-Target Risks
The advent of programmable gene editing technologies has revolutionized molecular biology and therapeutic development. Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and CRISPR-Cas systems represent three generations of gene editing tools, each with distinct mechanisms and specificity profiles [65] [2]. While ZFNs and TALENs rely on protein-DNA interactions for target recognition, CRISPR systems utilize a guide RNA (gRNA) to direct Cas nucleases to specific DNA sequences [2]. This fundamental difference has significant implications for specificity and ease of design. Although CRISPR offers unparalleled simplicity and versatility, its tendency for off-target effects—unintended edits at genomic sites with sequence similarity to the target—remains a primary concern for therapeutic applications [66] [67].
Artificial intelligence (AI) and machine learning (ML) are now addressing these critical limitations by bringing predictive accuracy to gRNA design and risk assessment. By analyzing complex sequence patterns and epigenetic contexts that influence editing outcomes, AI models are transforming CRISPR from a powerful but imperfect tool into a precise therapeutic modality [65] [68]. This technological convergence is particularly significant when contextualized against the more established specificity profiles of ZFNs and TALENs, which utilize protein-DNA recognition and often demonstrate high specificity at the cost of design complexity and limited scalability [8] [11].
Comparative Analysis of Gene Editing Platforms
The table below summarizes the key characteristics of ZFNs, TALENs, and CRISPR-Cas systems, highlighting their fundamental differences in design, specificity, and suitability for high-throughput applications.
Table 1: Comparison of Major Gene Editing Platforms
| Feature | ZFNs | TALENs | CRISPR-Cas Systems |
|---|---|---|---|
| Target Recognition | Protein-DNA [2] | Protein-DNA [2] | RNA-DNA [2] |
| Design Complexity | High (requires protein engineering) [8] | High (requires protein engineering) [8] | Low (only gRNA sequence needs changing) [8] [11] |
| Target Site Length | 18-36 bp/ZFN pair [2] | 30-40 bp/TALEN pair [2] | ~20 bp + PAM [2] |
| Multiplexing Potential | Low [2] | Low [2] | High [8] [2] |
| Scalability for High-Throughput | Challenging [2] | Challenging [2] | Excellent [8] |
| Reported Editing Efficiency | 0-12% [2] | 0-76% [2] | 0-81% [2] |
| Primary Specificity Concern | Less predictable off-target effects [2] | Less predictable off-target effects [2] | Predictable but frequent off-target effects [2] |
| Cost | High [8] [2] | High [8] [2] | Low [8] [2] |
CRISPR's primary advantage lies in its simplicity: redirecting to new genomic targets requires only the synthesis of a new gRNA, unlike ZFNs and TALENs which need laborious protein re-engineering [8] [2]. However, the RNA-guided DNA recognition mechanism of CRISPR can tolerate mismatches, particularly in the PAM-distal region, leading to potential off-target activity [67]. While ZFNs and TALENs are generally considered highly specific due to their longer recognition sequences and protein-DNA interaction mechanics, their off-target effects are noted to be "less predictable" compared to CRISPR [2]. This predictability in CRISPR's off-target profile is what makes it particularly amenable to AI-driven mitigation strategies.
AI and ML Tools for Enhancing CRISPR Specificity
AI models, particularly deep learning, have demonstrated remarkable success in predicting both gRNA on-target efficiency and off-target activity by learning from large-scale experimental datasets [65] [68]. The table below summarizes key AI tools and their applications in CRISPR research.
Table 2: AI/ML Models for gRNA Efficiency and Off-Target Prediction
| Tool/Model | AI Approach | Primary Application | Key Features |
|---|---|---|---|
| DeepCRISPR | Deep Learning [65] [68] | gRNA prediction & off-target activity [65] [68] | Integrates genomic context and sequence features [65] |
| CRISPick (Rule Set 3) | Light Gradient Boosting Machine (LightGBM) [68] | gRNA prediction [68] | Continuously updated model for SpCas9 [68] |
| DNABERT-Epi | Pre-trained DNA Foundation Model + Epigenetic Features [66] | Off-target prediction [66] | Integrates DNA sequence with epigenetic marks (H3K4me3, H3K27ac) [66] |
| CCLMoff | Transformer-based Language Model [69] [67] | Off-target prediction [69] [67] | Pre-trained on RNAcentral; strong generalization to unseen gRNAs [67] |
| SPROUT | Machine Learning [65] | Prediction of repair outcomes [65] | Predicts editing outcomes in primary T cells [65] |
These tools exemplify different strategic approaches. While earlier models like DeepCRISPR and CRISPick relied on supervised learning trained on specific CRISPR datasets, newer frameworks like DNABERT and CCLMoff leverage pre-trained foundation models [66] [67]. These foundation models are first trained on vast corpora of biological sequences (e.g., entire human genomes or RNA databases), allowing them to learn fundamental rules of nucleic acid architecture before being fine-tuned for the specific task of off-target prediction. This enables superior generalization, especially when encountering novel gRNA sequences not present in the training data [69].
Furthermore, the integration of epigenetic features such as histone modifications (H3K4me3, H3K27ac) and chromatin accessibility data (from ATAC-seq) provides a biological context that pure sequence-based models lack, significantly enhancing predictive accuracy [66]. The workflow below illustrates how these diverse data types are integrated in a modern AI framework for off-target prediction.
Experimental Protocols for Model Training and Validation
The development of robust AI models for CRISPR off-target prediction relies on standardized experimental and computational workflows. Below is a detailed methodology based on recent state-of-the-art approaches.
Data Curation and Preprocessing
The first critical step involves compiling a comprehensive dataset of validated on-target and off-target sites. For a tool like CCLMoff, this entails integrating data from multiple genome-wide detection techniques [67]:
- DSB Detection Methods: CIRCLE-seq, DISCOVER-seq, CHANGE-seq.
- Repair Product Detection Methods: GUIDE-seq, Digenome-seq, IDLV.
- Cas9 Binding Detection Methods: SITE-seq, Extru-seq.
Positive off-target sites are those validated by these experimental methods. To generate negative samples for training, tools like Cas-OFFinder are often used to scan the genome for sequences with high similarity to the gRNA but which are not confirmed off-targets, typically allowing for up to 6 mismatches and/or 1 bulge [67]. This creates a challenging negative set that helps the model learn to distinguish true off-targets from near-candidates.
Model Architecture and Training
The CCLMoff framework exemplifies a modern architecture. It treats off-target prediction as a question-answering task: the gRNA sequence is the "question" and a candidate DNA site is the "answer" [67].
- Input Representation: The gRNA and candidate DNA sequences are tokenized at the nucleotide level. The DNA sequence is converted to pseudo-RNA by replacing thymine (T) with uracil (U) to align with the pre-trained language model.
- Encoder: A transformer-based encoder, pre-trained on 23 million RNA sequences from RNAcentral (the RNA-FM model), processes the tokenized sequences. This transfer learning approach allows the model to incorporate fundamental knowledge of RNA secondary structure and interactions.
- Integration of Epigenetics (Optional): In an enhanced model like DNABERT-Epi or CCLMoff-Epi, epigenetic features are encoded using a Convolutional Neural Network (CNN) and then concatenated with the sequence representation from the language model [66] [67].
- Classification: The final hidden state corresponding to a special
[CLS]token is fed into a Multilayer Perceptron (MLP) that outputs a probability score for the candidate site being an off-target.
The model is trained using binary cross-entropy loss to minimize the difference between its predictions and the experimental labels [67]. The following diagram visualizes this integrated experimental and computational workflow.
Benchmarking and Interpretation
Rigorous benchmarking against existing methods (e.g., Cas-OFFinder, CRISPR-Net) across multiple independent datasets is essential [66] [67]. Performance is typically measured using metrics like AUC (Area Under the ROC Curve) and AUPRC (Area Under the Precision-Recall Curve). To move beyond "black box" predictions, interpretability techniques such as SHAP (SHapley Additive exPlanations) and Integrated Gradients are applied. These methods help identify which nucleotide positions in the gRNA:DNA duplex or which epigenetic features most strongly influenced the model's prediction, often validating known biological principles such as the critical importance of the seed region near the PAM site [66].
Successful implementation of AI-predicted gRNAs requires a suite of experimental and computational reagents. The table below details key solutions for validation and application.
Table 3: Essential Research Reagents and Resources for AI-Guided CRISPR Workflows
| Reagent/Resource | Function | Example Tools / Assays |
|---|---|---|
| gRNA Design Tools | Predicts optimal gRNA sequences for a target, incorporating on-target efficiency scores. | CRISPick [68], DeepCRISPR [65] [68] |
| Off-Target Prediction Software | Genome-wide scanning for potential off-target sites based on sequence similarity and AI models. | CCLMoff [67], DNABERT-Epi [66], Cas-OFFinder [67] |
| Genome-Wide Off-Target Detection Kits | Experimental validation of off-target effects using high-throughput sequencing. | GUIDE-seq [67], CIRCLE-seq [67], Digenome-seq [67] |
| Epigenomic Profiling Reagents | Assays to map chromatin features (accessibility, histone marks) for integration with AI models. | ATAC-seq, ChIP-seq for H3K4me3/H3K27ac [66] |
| Pre-trained Language Models | Foundation models providing sequence context for transfer learning in custom AI tool development. | RNA-FM [67], DNABERT [66] |
| Delivery Vectors | Efficient transport of CRISPR components into target cells for functional validation. | Adenovirus (AVV), Lentivirus [2] |
The integration of AI and machine learning with CRISPR technology marks a significant leap toward achieving the precision required for safe therapeutic genome editing. AI models have evolved from simple predictors to sophisticated tools that leverage pre-trained language models and multi-modal data, including epigenetic context, to forecast gRNA efficiency and off-target risks with growing accuracy [66] [68] [67]. This progress directly addresses CRISPR's historical specificity challenge, narrowing the gap with the high inherent specificity of older platforms like TALENs and ZFNs, but with far greater ease of design and scalability [8] [2].
As these AI tools become more interpretable and are trained on ever-larger and more diverse datasets, they pave the way for end-to-end gRNA design platforms. These platforms will be indispensable for researchers and drug development professionals aiming to design CRISPR-based therapies with minimized off-target risks, ultimately accelerating the translation of gene editing from the laboratory to the clinic.
Head-to-Head Comparison: A Decision Matrix for Tool Selection
The advent of programmable nucleases has revolutionized genetic engineering, offering unprecedented control over the genome. However, the potential for these powerful tools to cleave unintended genomic sites—a phenomenon known as off-target activity—remains a critical concern for their therapeutic and research applications. This guide provides a systematic comparison of the off-target specificities of the three primary genome-editing platforms: Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the CRISPR-Cas9 system. For researchers and drug development professionals, understanding the quantitative off-target profiles and the methodologies used to assess them is paramount for selecting the right tool for precise genetic interventions.
The fundamental difference in how these three technologies recognize their DNA targets underpins their varying specificity profiles.
Zinc Finger Nucleases (ZFNs) are fusion proteins comprising an array of zinc finger domains (each recognizing a 3-base pair DNA triplet) linked to the FokI nuclease domain. A pair of ZFNs must bind to opposite DNA strands, and their FokI domains must dimerize to create a double-strand break (DSB). This dimerization requirement inherently increases specificity, but the design is complex, as zinc fingers in an array can influence neighbors' specificity [19] [70] [71].
Transcription Activator-Like Effector Nucleases (TALENs) operate on a similar protein-DNA recognition principle. They fuse TAL effector (TALE) repeats (each repeat binds to a single nucleotide) to the FokI nuclease. Like ZFNs, they function in pairs, requiring dimerization for cleavage. A key advantage is that each TALE domain's binding is independent, making TALENs easier to design for high specificity with potentially lower off-target effects than ZFNs [19] [70] [71].
CRISPR-Cas9 employs a distinct, RNA-guided mechanism. The Cas9 nuclease is directed to its target DNA by a guide RNA (gRNA) through Watson-Crick base pairing. The target site must be adjacent to a short Protospacer Adjacent Motif (PAM). This system's simplicity is its strength; designing a new gRNA is far easier than engineering new proteins. However, Cas9 can tolerate mismatches and bulges between the gRNA and DNA, leading to a higher propensity for off-target effects compared to the protein-based systems [72] [70] [71].
Comparative Off-Target Rate Analysis: Quantitative Data
The following table synthesizes findings from peer-reviewed studies that directly or indirectly compare the off-target activities of these platforms.
Table 1: Comparative Off-Target Profiles of ZFNs, TALENs, and CRISPR-Cas9
| Editing Tool | Reported Off-Target Findings | Comparative Context | Key Study/Model System |
|---|---|---|---|
| Zinc Finger Nucleases (ZFNs) | Off-target mutations identified at 1 out of 10 pre-selected sites in 184 human pluripotent stem cell clones [19]. | In a head-to-head comparison targeting the CCR5 gene, ZFNs showed more off-target mutagenesis and greater cell toxicity than TALENs [19]. | Human pluripotent stem cells [19]; Human tumor cell lines [19]. |
| TALENs | Low but measurable mutagenesis rates at some of 19 potential off-target sites in human pluripotent stem cells [19]. | Demonstrated significantly fewer off-target mutations and lower cell toxicity than ZFNs when targeting the same site in the CCR5 gene [19]. | Human pluripotent stem cells [19]; Rice plants [10]. |
| CRISPR-Cas9 | Off-target effects are a well-documented challenge; unintended cleavage can occur at sites with partial gRNA complementarity [72] [50]. | Newer studies reveal risks beyond simple indels, including large structural variations (SVs) like kilobase- to megabase-scale deletions and chromosomal translocations, particularly when NHEJ is inhibited [50]. | Multiple human cell types; Comprehensive deep learning models benchmarked on NGS-based datasets [72] [73] [50]. |
Key Experimental Protocols for Off-Target Assessment
Robust assessment of off-target activity is critical. The following are key methodologies cited in the literature.
GUIDE-seq (Discovery of Off-Target Sites)
Purpose: To identify potential off-target sites of CRISPR-Cas9 nucleases in a genome-wide, unbiased manner in cellula [73]. Workflow:
- Transfection: Co-deliver CRISPR-Cas9 components (e.g., Cas9 and sgRNA expression plasmids) with a short, double-stranded oligodeoxynucleotide (dsODN) into cells.
- Integration: When a DSB occurs (either on-target or off-target), the dsODN is integrated into the break site via NHEJ.
- Genomic DNA Extraction & Sequencing: Harvest genomic DNA and perform high-throughput sequencing (e.g., next-generation sequencing).
- Bioinformatic Analysis: Map all sequencing reads containing the integrated dsODN sequence back to the reference genome to identify the genomic locations of DSBs.
CHANGE-seq (In Vitro Off-Target Profiling)
Purpose: A high-throughput in vitro method to map the off-target landscape of CRISPR-Cas9 [72]. Workflow:
- Library Preparation: Create a sequencing library from a reference human genomic DNA sample.
- In Vitro Cleavage: Incubate the library with the Cas9 nuclease complexed with its sgRNA (ribonucleoprotein, RNP).
- Adapter Ligation: Use a specialized adapter ligation strategy to specifically tag DNA ends generated by Cas9 cleavage.
- Sequencing & Analysis: Sequence the resulting fragments and bioinformatically identify all cleavage sites across the genome.
Deep Learning Models for Off-Target Prediction
Purpose: To computationally predict the likelihood of off-target cleavage for a given sgRNA, leveraging large datasets from methods like GUIDE-seq and CHANGE-seq. Protocol (as in DNABERT-Epi [72]):
- Data Acquisition & Preprocessing: Curate and pre-process multiple off-target datasets. Address severe class imbalance (many more negative sites than positive) through techniques like random downsampling.
- Feature Integration: For a given sgRNA and a candidate off-target DNA sequence, the model analyzes:
- Sequence Context: Using a pre-trained DNA language model (DNABERT) that understands genomic sequence patterns.
- Epigenetic Features: Incorporate cell-specific epigenetic data (e.g., chromatin accessibility ATAC-seq, histone marks H3K4me3 and H3K27ac) as normalized signal vectors within a window around the candidate site.
- Model Training & Validation: Train the model to distinguish active from inactive off-target sites. Performance is rigorously evaluated using cross-validation and on independent test datasets.
The workflow for integrating these methods into a comprehensive off-target assessment strategy is illustrated below.
Table 2: Key Research Reagent Solutions for Off-Target Analysis
| Reagent / Resource | Function in Off-Target Assessment | Specific Examples / Notes |
|---|---|---|
| Programmable Nuclease Kits | Provides the core editing machinery (ZFNs, TALENs, or CRISPR-Cas9) for inducing DSBs. | Commercial ZFN and TALEN pairs; CRISPR-Cas9 systems (e.g., wild-type, high-fidelity variants like HiFi Cas9 [50]). |
| Off-Target Detection Kits | Streamlined reagents for experimental off-target identification. | GUIDE-seq kits [73]; CIRCLE-seq kits [73]. |
| Validated Cell Lines | Provide a consistent biological model for evaluating editing efficiency and specificity. | HEK293T, U2OS, and CD4+/CD8+ T-cells are commonly used in off-target studies [72]. |
| Epigenetic Data | Provides cell-specific information on chromatin state, which influences Cas9 accessibility. | Publicly available from GEO (e.g., ATAC-seq, H3K4me3, H3K27ac ChIP-seq data) [72]. |
| Computational Tools & Software | Predict potential off-target sites in silico to guide experimental design. | CCTop, Cas-OFFinder [73]; Advanced deep learning models like DNABERT-Epi [72] and CCLMoff [73]. |
The direct comparison of ZFNs, TALENs, and CRISPR-Cas9 reveals a classic trade-off between ease-of-use and intrinsic specificity. TALENs consistently demonstrate high specificity and low off-target rates, making them a strong candidate for applications where precision is paramount and the higher cost and complexity of protein engineering are justifiable [19] [71]. CRISPR-Cas9, while the most versatile and user-friendly system, contends with a higher risk of off-target effects and newly appreciated on-target genomic aberrations like large structural variations [50].
The future of safe genome editing lies in continuous innovation. For CRISPR, this includes:
- Engineered High-Fidelity Cas Variants: Such as HiFi Cas9, designed to reduce off-target cleavage while maintaining on-target activity [50].
- Advanced Delivery Methods: Transient delivery of pre-assembled Cas9 ribonucleoprotein (RNP) complexes can shorten the editing window, reducing off-target opportunities [70].
- Sophisticated Computational Prediction: The integration of foundation models and epigenetic data, as seen with DNABERT-Epi, is becoming crucial for comprehensive pre-clinical safety profiling [72].
In conclusion, the choice of editing platform is context-dependent. For large-scale functional screens, CRISPR's scalability is unmatched. For clinical applications requiring the highest possible certainty of single-site precision, the proven track record of TALENs or the use of the most advanced, high-fidelity CRISPR systems is warranted. A thorough, multi-faceted off-target assessment, combining state-of-the-art computational prediction with rigorous experimental validation, remains the non-negotiable standard for any serious therapeutic or research application.
The advent of programmable gene-editing technologies has revolutionized genetic research and therapeutic development. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), Transcription Activator-Like Effector Nucleases (TALENs), and Zinc Finger Nucleases (ZFNs) represent three generations of these powerful tools. While often discussed in terms of precision, their performance in scalability—spanning high-throughput genetic screens to targeted, single-locus modifications—is equally critical for research and clinical applications. This guide objectively compares the efficiency and throughput of these platforms, providing a structured framework for selecting the appropriate tool based on project scale and specificity requirements.
Each gene-editing platform employs a distinct mechanism to achieve DNA cleavage, which fundamentally dictates its scalability and ease of use.
CRISPR-Cas9: This system utilizes a guide RNA (gRNA) to direct the Cas9 nuclease to a specific DNA sequence. The simple base-pairing rules of RNA-DNA binding mean that targeting a new genomic site only requires the synthesis of a new gRNA, a process that is both rapid and inexpensive [8]. This simplicity makes CRISPR exceptionally suited for multiplexing (editing multiple genes simultaneously) and large-scale library screens [10].
TALENs: TALENs are fusion proteins consisting of a Transcription Activator-Like Effector (TALE) DNA-binding domain and a FokI nuclease domain. Each TALE repeat recognizes a single DNA base pair. While this offers high specificity, constructing a new TALEN for each target requires time-intensive protein engineering, as the entire TALE array must be re-assembled [8] [3].
ZFNs: As the first generation of programmable nucleases, ZFNs link a Zinc Finger DNA-binding domain to a FokI nuclease. Each zinc finger module recognizes a DNA triplet, and multiple modules must be assembled to achieve specificity. The design is complicated by context-dependent effects, where the performance of one zinc finger can influence its neighbors. This makes ZFN design, validation, and synthesis a technically demanding and lengthy process [8] [11].
The core differentiator for scalability lies in the design paradigm: CRISPR's RNA-guided system separates the easily programmable targeting component (gRNA) from the effector (Cas9), whereas TALENs and ZFNs require the entire targeting moiety to be re-engineered at the protein level for each new target.
Quantitative Performance Comparison
Direct comparisons of efficiency, specificity, and scalability reveal clear trade-offs between these technologies, as summarized in the table below.
Table 1: Direct Comparison of Gene-Editing Platforms for Scalability and Efficiency
| Feature | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Targeting Molecule | Guide RNA (gRNA) | TALE Protein | Zinc Finger Protein |
| Ease of Design & Cloning | Very High (days) [8] | Moderate (weeks) [8] | Low (months) [8] [10] |
| Relative Cost | Low [8] | High [8] | Very High [8] |
| Suitability for High-Throughput Screening | Excellent [8] | Limited [8] | Poor [8] |
| Multiplexing (Editing Multiple Genes) | Excellent [8] | Difficult [3] | Difficult [3] |
| Editing Efficiency | High [13] | High [3] | Variable [3] |
| Reported Off-Target Effects (in HPV Model) | E6: 0, E7: 4, URR: 0 [13] | E6: 7, E7: 36 [13] | URR: 287 [13] |
The data from a direct comparative study using the GUIDE-seq method to target the Human Papillomavirus (HPV) genome is particularly telling. CRISPR-Cas9 demonstrated superior specificity, with zero off-target events detected in the E6 and URR regions, significantly fewer than TALENs and vastly fewer than ZFNs in the same loci [13]. This combination of high efficiency, superior specificity in some contexts, and simple design makes CRISPR the dominant tool for high-throughput applications.
Experimental Protocols for Assessing Efficiency and Specificity
Robust experimental protocols are essential for quantitatively comparing the performance of these editing tools. The following workflow and a cited example from the literature provide a standard approach.
Diagram 1: Workflow for Editing Tool Comparison
Detailed Methodology: GUIDE-seq for Off-Target Profiling
A seminal study directly compared ZFNs, TALENs, and SpCas9 using a standardized protocol focused on HPV-targeted therapy [13].
- 1. Target Selection: The study targeted three key regions of the HPV16 genome: the upstream regulatory region (URR) and the E6 and E7 oncogenes.
- 2. Reagent Design and Delivery:
- ZFNs & TALENs: Multiple designs were generated for each target. For ZFNs, specificity was found to be inversely correlated with the count of middle "G" in zinc finger proteins. For TALENs, different N-terminal domains (WT/αN/βN) and recognition modules (NN/NH) were tested [13].
- CRISPR-Cas9: gRNAs were designed to target the same HPV16 sequences.
- All nucleases were delivered into human cells using plasmid transfection.
- 3. Off-Target Detection via GUIDE-seq: Following transfection, the GUIDE-seq method was employed. This involves:
- Introducing a blunt, double-stranded oligodeoxynucleotide tag into double-strand breaks (DSBs) generated by the nucleases.
- Enriching and sequencing the tagged DNA fragments.
- Mapping all DSB sites across the genome using a novel bioinformatics pipeline developed for ZFNs and TALENs to allow a parallel comparison [13].
- 4. Data Analysis: The number of unique off-target sites was quantified for each nuclease at each locus. The results conclusively showed that SpCas9 was more efficient and specific than the ZFNs and TALENs tested, with fewer off-target counts across all targeted regions [13].
The Scientist's Toolkit: Essential Reagents for Gene-Editing Comparisons
Successful execution of gene-editing experiments requires a suite of specialized reagents and tools.
Table 2: Key Research Reagent Solutions for Gene-Editing Studies
| Reagent / Solution | Function | Example Use-Case |
|---|---|---|
| Guide RNA (gRNA) Libraries | Synthetic RNA pools for targeted gene knockout or activation in high-throughput CRISPR screens. | Genome-wide loss-of-function screens to identify essential genes for drug discovery [8]. |
| TALEN Repeat Kits | Modular protein assembly kits for constructing TALE DNA-binding domains. | Creating precise edits in cell lines for stable cell line generation where CRISPR off-targets are a concern [8]. |
| Zinc Finger Fusion Plasmids | Pre-designed plasmids for expressing ZFN pairs with FokI nuclease. | Targeted gene correction in therapeutic applications with a long history of validation [3]. |
| GUIDE-seq Oligos | Double-stranded oligodeoxynucleotide tags for genome-wide identification of nuclease off-target activity. | Profiling and comparing the specificity of ZFNs, TALENs, and CRISPR-Cas9 in any cell type [13]. |
| Liquid Handling Systems | Automated instruments for precise dispensing of small liquid volumes in microplates. | Essential for high-throughput screening workflows to ensure consistency and efficiency [74]. |
| Multimode Plate Readers | Instruments for detecting biological signals (e.g., fluorescence, luminescence) in assay plates. | Enabling high-content phenotypic assays in high-throughput drug discovery campaigns [75]. |
Application Contexts: Choosing the Right Tool
The choice between these platforms is context-dependent, hinging on the core trade-off between throughput and validated precision.
For High-Throughput Functional Genomics: CRISPR is Unmatched CRISPR screening is the established standard for large-scale functional genomics. Its ability to be used in pooled or arrayed formats with thousands to hundreds of thousands of unique gRNAs allows researchers to systematically knock out or activate genes across the entire genome [8]. This is invaluable for identifying novel drug targets, understanding genetic dependencies, and uncovering resistance mechanisms in oncology and beyond. The simplicity of gRNA synthesis makes these massive screens feasible and cost-effective.
For Targeted Modifications with Validated Precision: A Nuanced Choice
- TALENs are often preferred for projects requiring high specificity in complex genomic regions, such as those with high GC content or repetitive sequences, where CRISPR's off-target activity may be a greater concern [3] [11]. Their protein-based targeting can offer a more predictable off-target profile in certain contexts.
- ZFNs, while historically important, are now typically reserved for niche applications where a specific ZFN has already been extensively validated and possesses a known, acceptable safety profile, such as in some legacy clinical-grade therapies [8].
Table 3: Technology Selection Guide Based on Application
| Application Goal | Recommended Tool | Rationale |
|---|---|---|
| Genome-wide Loss-of-Function Screen | CRISPR-Cas9 | Unparalleled scalability and cost-effectiveness for designing and deploying large gRNA libraries [8]. |
| Multiplexed Gene Knock-in | CRISPR-Cas9 | Ability to co-deliver multiple gRNAs and donor templates for simultaneous edits at different loci [8]. |
| Precise Editing in a Challenging Locus | TALENs | High protein-driven specificity can be advantageous for regions where CRISPR gRNAs perform poorly [3] [11]. |
| Clinical Application with a Well-Characterized ZFN | ZFNs | Leverages existing preclinical and regulatory validation for that specific therapeutic construct [8]. |
The scalability of gene-editing technologies presents a clear trade-off. CRISPR-Cas9 stands out as the superior platform for high-throughput screens due to its simple design, low cost, and exceptional multiplexing capability. In contrast, TALENs remain a competitive option for targeted modifications demanding the highest possible specificity, particularly in niche contexts where their more complex design is a justifiable investment. ZFNs, as the pioneering technology, are increasingly specialized for specific, validated applications.
The ongoing evolution of CRISPR, including the development of high-fidelity Cas9 variants, base editors, and prime editors, continues to blur the lines by enhancing its precision without sacrificing scalability. For most modern research and drug discovery programs focused on throughput, CRISPR-Cas9 is the unequivocal tool of choice.
The advent of programmable nucleases has revolutionized genetic engineering, offering researchers unprecedented control over genomic sequences. Among these tools, Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and CRISPR-Cas9 represent three generations of genome editing technology, each with distinct implications for project design, cost, and resource allocation. For researchers and drug development professionals, the choice between these systems is not merely scientific but also a strategic decision influenced by technical feasibility, timelines, and budgetary constraints. This guide provides an objective comparison of these platforms, focusing on their ease of use, development timelines, and the resource investment required for their implementation, thereby equipping scientists with the data needed for efficient project planning.
Comparative Analysis of Design and Development Parameters
The design and construction of these gene-editing tools involve varying levels of complexity, which directly impact project timelines and required expertise. The following table summarizes the key parameters that influence resource allocation.
Table 1: Design, Timeline, and Cost Comparison of Gene-Editing Technologies
| Feature | ZFNs | TALENs | CRISPR-Cas9 |
|---|---|---|---|
| Mechanism of Target Recognition | Protein-DNA interaction [1] [76] | Protein-DNA interaction [1] [76] | RNA-DNA interaction (Watson-Crick base pairing) [8] [76] |
| Design Complexity | High; challenging due to context-dependent effects between fingers [1] [10] | Moderate; modular but repetitive assembly [8] [10] | Low; simple guide RNA (gRNA) design [8] [11] |
| Development Timeline | Several weeks to months [10] | Several days to weeks [10] | A few days [8] |
| Relative Cost | High [8] [77] | High [77] | Low; 3-6 fold cheaper per reaction than TALENs [77] |
| Key Design Constraint | Target sequence size and context [1] [10] | Large cDNA size (~2kb larger than ZFNs) complicating delivery [10] | Requirement for a Protospacer Adjacent Motif (PAM) sequence [10] [76] |
Analysis of Design Workflows
The journey from target sequence to a functional gene-editing tool differs significantly across platforms. ZFNs require the engineering of a protein domain where each zinc finger recognizes a DNA triplet. A major hurdle is the "context-dependent" interaction between neighboring fingers, making the design process complex and often requiring sophisticated selection methods like Oligomerized Pool Engineering (OPEN) to achieve functional arrays [1]. This technical challenge is a primary reason why ZFN development can take months and is often considered the most expertise-intensive of the three [10].
TALENs simplified the design paradigm by introducing a one-to-one code: a single TALE repeat domain recognizes a single DNA base pair. This modularity makes their design more straightforward than ZFNs [1]. However, the cloning process remains laborious due to the highly repetitive nature of the TALE sequences, requiring specialized assembly methods like "Golden Gate" cloning [1]. Furthermore, the large size of TALEN-encoding DNA (its cDNA is typically 2 kb larger than that of a ZFN) can pose a challenge for delivery into cells using certain viral vectors, impacting experimental planning [10].
In contrast, the CRISPR-Cas9 system decouples the targeting component from the nuclease. Target recognition is achieved by a short guide RNA (gRNA) that is complementary to the DNA sequence of interest. Designing a new target requires only the synthesis of a new ~20 nucleotide gRNA sequence, a process that is fast, inexpensive, and does not require complex protein engineering [8] [11]. This fundamental difference is why new CRISPR-Cas9 targets can be designed and tested in a matter of days, democratizing access to gene editing for labs without specialized protein engineering expertise [8].
The workflow from sequence selection to functional nuclease validation highlights these fundamental differences in complexity and time investment.
Experimental Protocols for Specificity and Efficiency Assessment
A critical step in any gene-editing project is the empirical validation of the designed nucleases. Key performance metrics include on-target efficiency and off-target activity. Below is a detailed protocol for a widely used method to assess genome-wide off-target effects.
GUIDE-seq (Genome-Wide Unidentified Double-Strand Break Enabled by Sequencing)
Principle: This unbiased method detects double-strand breaks (DSBs) in vivo by capturing the integration sites of a transfected double-stranded oligodeoxynucleotide (dsODN) tag. These tags serve as markers for sequencing and genome-wide off-target identification [60].
Key Reagents and Functions:
- Programmed Nuclease: The ZFN, TALEN, or CRISPR-Cas9 construct to be tested.
- dsODN Tag: A short, double-stranded DNA oligo that integrates into DSB sites during repair, acting as a molecular "tag" for the break [60].
- T7 Endonuclease I (T7E1): An enzyme used for initial, rapid assessment of nuclease activity by cleaving heteroduplex DNA formed at indel mutation sites.
- PCR Reagents: For amplifying the regions flanking the integrated dsODN tag.
- Next-Generation Sequencing (NGS) Platform: For high-throughput sequencing of PCR-amplified fragments to map all dsODN integration sites across the genome.
Procedure:
- Co-transfection: Co-transfect the target cells with the plasmids encoding the nuclease (e.g., ZFN, TALEN, or CRISPR-Cas9 + gRNA) and the dsODN tag.
- Incubation: Allow cells to grow for a sufficient period (e.g., 72-96 hours) for nuclease cleavage and dsODN tag integration to occur.
- Genomic DNA Extraction: Harvest cells and isolate high-quality genomic DNA.
- dsODN Breakpoint PCR: Perform PCR using one primer specific to the integrated dsODN tag and another primer specific to a known on-target site. This serves as a quality control to confirm successful tag integration at the intended target [60].
- GUIDE-seq Library Construction: Fragment the genomic DNA and use capture primers specific to the dsODN tag to enrich for fragments containing the integration sites. Prepare these fragments for NGS.
- Sequencing and Bioinformatics Analysis: Sequence the library on an NGS platform. Use specialized bioinformatics pipelines to align the sequences to the reference genome and identify all locations where the dsODN tag has integrated, which correspond to both on-target and off-target DSB events [60].
The following diagram illustrates the core workflow of the GUIDE-seq protocol, from tag integration to off-target analysis.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of gene-editing experiments requires a suite of reliable reagents. The table below details essential materials and their functions for developing and validating programmable nucleases.
Table 2: Key Research Reagent Solutions for Gene Editing
| Reagent / Material | Function in Experimentation |
|---|---|
| Pre-designed ZFN/TALEN Libraries | Commercial libraries (e.g., CompoZr ZFNs) can bypass the need for in-house design and validation, saving time but at a higher cost [1]. |
| TALE Assembly Kits | Kits employing Golden Gate or other cloning methods are essential for efficient and accurate assembly of repetitive TALEN arrays [1]. |
| Cas9 Expression Vectors | Ready-to-use plasmids encoding Cas9 nuclease (wild-type, nickase, or dead variants) form the backbone of CRISPR experiments [76]. |
| gRNA Cloning Vectors | Plasmids designed for easy insertion of a ~20nt target sequence for gRNA expression [76]. |
| Delivery Vehicles (e.g., Viral Vectors) | Critical for transporting editing components into target cells. The large size of TALENs can be a constraint for some viral vectors [10]. |
| Validation Enzymes (e.g., T7E1) | Enzymes like T7 Endonuclease I are used for fast, initial screening of nuclease-induced indel mutations [60]. |
| dsODN Tag for GUIDE-seq | The core reagent for unbiased, genome-wide off-target detection using the GUIDE-seq method [60]. |
| NGS Kits and Platforms | Essential for comprehensive analysis of editing outcomes (on-target efficiency) and off-target profiling (e.g., via GUIDE-seq) [60]. |
The strategic choice between ZFNs, TALENs, and CRISPR-Cas9 involves a direct trade-off between design complexity, time, and cost. ZFNs, while pioneering, demand the most resources in terms of expert knowledge, time, and financial investment. TALENs offer a more predictable design code but share similar cost and delivery challenges. In contrast, CRISPR-Cas9 has dramatically lowered the barriers to entry through its simple, RNA-guided design, rapid turnaround times, and significantly lower cost.
For most research and drug development applications, CRISPR-Cas9 provides a superior balance of ease-of-use, speed, and cost-effectiveness, making it the preferred starting point for new projects. However, TALENs remain a valuable tool for applications requiring extreme specificity or targeting genomic regions inaccessible to standard CRISPR-Cas9 systems. Ultimately, aligning the technical strengths and resource demands of each platform with project-specific goals and constraints is key to successful and efficient genome engineering.
The advent of programmable gene-editing technologies has revolutionized molecular biology, providing researchers with an unprecedented ability to investigate gene function and develop therapeutic interventions for genetic disorders [8]. CRISPR-Cas9, TALENs (Transcription Activator-Like Effector Nucleases), and ZFNs (Zinc Finger Nucleases) represent three foundational technologies in this space, each with distinct mechanisms and applications [45] [78]. While ZFNs pioneered targeted genome editing as the first programmable nucleases, TALENs emerged with simplified design, and CRISPR-Cas9 has dramatically accelerated adoption through its RNA-guided simplicity [79]. This guide provides an objective comparison of these platforms, synthesizing experimental data and technical specifications to empower researchers in selecting the optimal technology for specific applications in research and drug development.
Molecular Mechanisms and Technical Specifications
Fundamental Operating Principles
Each editing platform employs a distinct DNA recognition and cleavage mechanism, which fundamentally influences its ease of use, specificity, and application potential.
ZFNs utilize engineered zinc finger proteins, where each finger recognizes a 3-base pair DNA triplet, fused to the FokI nuclease domain [45] [78]. Effective DNA cleavage requires two ZFN monomers binding to opposite DNA strands with precise spacing and FokI dimerization [78].
TALENs similarly employ the FokI nuclease but use TALE (Transcription Activator-Like Effector) proteins for DNA recognition, where each single TALE repeat recognizes one specific nucleotide [45] [10]. This one-to-one correspondence simplifies design compared to ZFNs [10].
CRISPR-Cas9 operates through a guide RNA (gRNA) that directs the Cas9 nuclease to complementary DNA sequences via Watson-Crick base pairing [8] [45]. Target recognition requires a Protospacer Adjacent Motif (PAM) sequence adjacent to the target site [10].
Comparative Technical Specifications
Table 1: Technical comparison of major gene-editing platforms
| Feature | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Recognition Mechanism | RNA-DNA hybridization [78] | Protein-DNA (TALE repeats) [78] | Protein-DNA (Zinc fingers) [78] |
| DNA Recognition Length | 20bp + PAM sequence [78] | 30-40bp [78] | 9-18bp [78] |
| Nuclease Component | Cas9 [8] | FokI (requires dimerization) [78] | FokI (requires dimerization) [78] |
| Targeting Efficiency | High (up to 70% success rate in random DNA) [77] | High (96% affinity rate) [10] | Moderate [8] |
| Multiplexing Capability | High (multiple gRNAs) [8] | Limited [8] | Limited [8] |
| Primary Repair Mechanism | NHEJ/HDR [8] | NHEJ/HDR [10] | HDR [10] |
Performance Comparison and Experimental Data
Efficiency, Specificity, and Practical Considerations
Quantitative assessment of editing performance reveals critical trade-offs between efficiency, specificity, and practical implementation factors that influence technology selection.
Editing Efficiency and Success Rates: CRISPR-Cas9 demonstrates approximately 70% probability of successfully targeting random DNA sequences, while TALENs achieve DNA binding affinity rates up to 96% in optimal conditions [10] [77]. ZFNs show more variable efficiency dependent on zinc finger array optimization [8].
Off-Target Effects: Protein-based systems (TALENs and ZFNs) generally exhibit lower off-target effects due to the requirement for nuclease dimerization and more stringent DNA recognition [11] [78]. CRISPR's off-target activity stems from gRNA binding to partially complementary sequences, though high-fidelity Cas9 variants (e.g., HF-Cas9, eCas9) have significantly improved specificity [78].
Experimental Workflows and Timelines: CRISPR system design can be completed within days, while TALEN and ZFN construction typically requires weeks to months of protein engineering [8] [45]. This dramatic difference in implementation timeline has accelerated CRISPR adoption significantly [79].
Table 2: Experimental performance and practical implementation factors
| Parameter | CRISPR-Cas9 | TALENs | ZFNs |
|---|---|---|---|
| Design Complexity | Low (guide RNA design) [8] | Medium (protein engineering) [8] | High (complex protein design) [8] |
| Development Timeline | Days [8] | Weeks [10] | Months [10] |
| Relative Cost | Low [8] | Medium [8] | High [8] |
| Off-Target Risk | Moderate to High [11] | Low [11] | Low to Moderate [8] |
| Cell Toxicity | Low [8] | Low [10] | Moderate [10] |
| Delivery Efficiency | High (multiple formats) [8] | Challenging (large size) [10] | Moderate [45] |
Experimental Design and Validation Workflow
Decision Matrix for Technology Selection
Application-Based Technology Recommendations
The optimal gene-editing technology varies significantly based on project requirements, with considerations for precision, scale, and resource constraints guiding selection.
High-Throughput Functional Genomics: CRISPR is the unequivocal choice for genome-wide screens and multiplexed editing applications due to its scalability and simplified design [8]. Large-scale CRISPR screening enables systematic identification of essential genes and novel drug targets with unprecedented efficiency [8].
Therapeutic Applications Requiring High Specificity: For clinical applications where off-target effects present significant safety concerns, TALENs provide superior specificity and are established in therapies for conditions like HIV and hemophilia [8]. ZFNs also maintain relevance in validated clinical contexts with proven precision [8].
Complex Loci and Challenging Targets: TALENs demonstrate advantages for editing repetitive sequences or regions with high GC content where CRISPR efficiency may be compromised [11]. Their modular DNA recognition provides flexibility for difficult genomic contexts.
Resource-Constrained Environments: CRISPR's dramatically lower costs and simplified implementation make it accessible for academic labs and projects with budget limitations [8] [77]. The technology has democratized gene editing by reducing barriers to entry [8].
Table 3: Application-based technology selection matrix
| Application Scenario | Recommended Technology | Rationale | Key Considerations |
|---|---|---|---|
| Large-Scale Genetic Screens | CRISPR-Cas9 | Unparalleled scalability and multiplexing capability [8] | Monitor for off-target effects using validated controls [80] |
| Clinical Therapeutic Development | TALENs/ZFNs | Proven high specificity and regulatory familiarity [8] | Higher development costs and longer timelines [8] |
| Rapid Prototyping/Proof-of-Concept | CRISPR-Cas9 | Fast design and low cost [8] | Ideal for initial validation before moving to more specific platforms |
| Editing High-GC/Repetitive Regions | TALENs | Superior performance in challenging genomic contexts [11] | More complex delivery due to larger size [10] |
| Base Editing (No DSBs) | CRISPR-Derived Base Editors | Enables precise single-nucleotide changes [44] [45] | Reduced off-target risks compared to standard CRISPR [44] |
| Large DNA Insertions | CRISPR with Recombinases/Transposases | Expanded size capabilities through fusion systems [44] | Emerging technology with optimization required |
Research Reagent Solutions and Essential Materials
Successful implementation of gene-editing technologies requires specific reagent systems and validation tools. The following table outlines core components for establishing editing workflows.
Table 4: Essential research reagents for gene-editing workflows
| Reagent Category | Specific Examples | Function | Technology Compatibility |
|---|---|---|---|
| Nuclease Components | SpCas9, SaCas9, FokI variants | Core editing enzymes that create DNA breaks [45] | Platform-specific |
| Targeting Molecules | gRNA expression vectors, TALE arrays, Zinc finger arrays | Sequence specificity determinants [78] | Platform-specific |
| Delivery Vehicles | AAV, Lentivirus, Lipid Nanoparticles | Intracellular delivery of editing components [8] | Cross-platform (with size considerations) |
| Validation Assays | T7E1 Surveyor, GUIDE-seq, Sanger sequencing | Detection and quantification of editing events and off-target effects [78] | Cross-platform |
| HDR Templates | Single-stranded DNA donors, Double-stranded DNA vectors | Template for precise edits via homologous recombination [8] | Cross-platform |
| Cell Culture Reagents | Transfection reagents, Selection antibiotics | Maintenance and selection of edited cells [8] | Cross-platform |
Emerging Trends and Future Perspectives
The gene-editing landscape continues to evolve rapidly with next-generation technologies addressing current limitations. Base editing and prime editing systems enable precise nucleotide changes without double-strand breaks, significantly reducing off-target risks [44] [45]. CRISPR-Cas variants with altered PAM requirements (e.g., Cas12, Cas13) expand targetable genomic space [8] [78]. Additionally, epigenetic editors capable of stable gene manipulation without permanent DNA changes represent another frontier [44]. The global genome editing market reflects this innovation trajectory, projected to grow from $4.25 billion in 2025 to $13.36 billion by 2035, with CRISPR technology capturing dominant market share [79]. As these technologies mature, context-specific optimization rather than universal superiority will guide researcher implementation, with each platform finding its niche in the expanding gene-editing toolkit.
Conclusion
The choice between CRISPR, TALENs, and ZFNs is not a declaration of a single winner but a strategic decision based on the specific requirements of the project. CRISPR offers unparalleled ease, scalability, and a rapidly evolving toolkit, making it the default for most high-throughput and therapeutic applications. TALENs remain indispensable for scenarios demanding the utmost protein-DNA driven specificity, such as mitochondrial genome editing or when CRISPR's PAM requirements are restrictive. ZFNs, while historically critical, see more limited use today. The future of gene editing specificity lies in the continued engineering of CRISPR systems—like base and prime editors that avoid double-strand breaks—and the powerful integration of AI to predict and eliminate off-target effects. For biomedical research, this means a growing arsenal of more precise, safer tools ready for clinical translation, ultimately enabling cures for a broader range of genetic diseases.
References
- 1. ZFN, TALEN and CRISPR/Cas-based methods for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3694601/]
- 2. Exploring CRISPR-Cas: The transformative impact of gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506487/]
- 3. CRISPR vs. TALEN vs. ZFN: Which Gene Editing Tool Is ... [https://synapse.patsnap.com/article/crispr-vs-talen-vs-zfn-which-gene-editing-tool-is-right]
- 4. Multi-targeting zinc finger nuclease vector unsilences ... [https://www.nature.com/articles/s41434-025-00582-1]
- 5. Optimized tuning of TALEN specificity using non- ... [https://www.nature.com/articles/srep08150]
- 6. Revolution of Biotechnology with CRISPR [https://www.nature.com/articles/s12276-025-01452-x]
- 7. CRISPR Delivery Systems for Organ-Specific Targeting [https://www.sciencedirect.com/science/article/pii/S2950482125000336]
- 8. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 9. Expanding the genetic editing tool kit: ZFNs, TALENs, and ... [https://www.jci.org/articles/view/72992]
- 10. A Comparative Review of Genetic Modification Tools for ... [https://nhsjs.com/2025/a-comparative-review-of-genetic-modification-tools-for-crop-improvement-efficiency-scalability-and-usability/]
- 11. CRISPR-Cas9 vs TALEN vs ZFN: Precision Gene Editing ... [https://synapse.patsnap.com/article/crispr-cas9-vs-talen-vs-zfn-precision-gene-editing-compared]
- 12. State of CRISPR Gene Editing in the Drug Discovery Sector [https://www.synthego.com/blog/survey-report-crispr-therapeutics/]
- 13. The comparison of ZFNs, TALENs, and SpCas9 by GUIDE- ... [https://www.sciencedirect.com/science/article/pii/S216225312100202X]
- 14. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOoqO_89oK5uWS0jUojNMKKOH90Y_2q4XYpy6LqcCWrbiHpSmjWWa]
- 15. Comparison of CRISPR/Cas9 and TALENs on editing an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4916124/]
- 16. GE: Which Should I Choose, TALEN or CRISPR [https://www.genecopoeia.com/resource/genome-editing-talen-or-crispr/]
- 17. What is the PAM in Sequence Experiments, and Why is it... CRISPR [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 18. Protospacer adjacent motif - Wikipedia [https://en.wikipedia.org/wiki/Protospacer_adjacent_motif]
- 19. Expanding the genetic editing tool kit: ZFNs, TALENs, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4191047/]
- 20. identification by PAM -Cas effector complexes: diversified... CRISPR [https://pmc.ncbi.nlm.nih.gov/articles/PMC6546366/]
- 21. Custom CRISPR–Cas9 PAM variants via scalable ... [https://www.nature.com/articles/s41586-025-09021-y]
- 22. Comparison of the editing patterns and editing efficiencies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5901495/]
- 23. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/]
- 24. Expanded activity of dimer nucleases by combining ZFN ... [https://www.nature.com/articles/srep02376]
- 25. Improved Specificity and Safety of Anti-Hepatitis B Virus TALENs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8310341/]
- 26. Advancing CRISPR genome editing into gene therapy clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12094669/]
- 27. FDA Approves First Gene Therapies to Treat Patients with ... [https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease]
- 28. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 29. Gene Editing as the Future of Cardiac Amyloidosis ... [https://www.sciencedirect.com/science/article/abs/pii/S0146280623001585]
- 30. Early Findings from Novel CRISPR Therapy Show Promise ... [https://arci.org/nobel-prize-winning-science-being-applied-in-attr-amyloidosis/]
- 31. CRISPR/Cas9 gene editing for curing sickle cell disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC8049447/]
- 32. Single AAV-mediated CRISPR-Nme2Cas9 efficiently ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8753293/]
- 33. TALENs—an indispensable tool in the era of CRISPR [https://pmc.ncbi.nlm.nih.gov/articles/PMC8380213/]
- 34. Powering new therapeutics with precision mitochondrial ... [https://www.nature.com/articles/s41587-025-02693-x]
- 35. Mitochondrial Base Editing of the m.8993T>G Mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12652715/]
- 36. Current Progress of Mitochondrial Genome Editing by ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2022.883459/full]
- 37. Broad Specificity Profiling of TALENs Results in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4010127/]
- 38. CRISPR Agriculture News Today: 7 Powerful Advances in ... [https://farmonaut.com/news/crispr-agriculture-news-today-7-powerful-advances-in-2025]
- 39. Synthetic biology and metabolic engineering strategies in ... [https://www.sciencedirect.com/science/article/pii/S0926669025006065]
- 40. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOorFmHOCD4qZ7fz6AwnEJOh1eBa5rCNr_HswQ4QbE0CT2QSO_EiN]
- 41. TALEN-Interceded Genome Editing in Plants [https://pubmed.ncbi.nlm.nih.gov/41095166/]
- 42. CRISPR in Agriculture Market Share, and Analysis 2025- ... [https://www.nextmsc.com/report/crispr-in-agriculture-market-3430]
- 43. The Strategy and Application of Gene Attenuation in Metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12029929/]
- 44. Recent advances in therapeutic gene-editing technologies [https://www.sciencedirect.com/science/article/pii/S152500162500200X]
- 45. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 46. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9176214/]
- 47. The game-changer in CRISPR-Cas9 genome editing [https://www.sciencedirect.com/science/article/pii/S2405844024006376]
- 48. Genome-wide Specificity of Highly Efficient TALENs and ... [https://www.sciencedirect.com/science/article/pii/S2329050117300098]
- 49. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 50. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 51. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 52. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 53. Off-target sequence variations driven by the intrinsic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12273960/]
- 54. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 55. Peeling back the layers of immunogenicity in Cas9-based ... [https://www.sciencedirect.com/science/article/pii/S1525001625005350]
- 56. Scientists engineer CRISPR enzymes that evade ... [https://www.broadinstitute.org/news/scientists-engineer-crispr-enzymes-evade-immune-system]
- 57. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 58. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOooQWbDpoC_c6tRbIMhPM_hae7idmjLd6_J2-dc2k4l23C7NH6ji]
- 59. Base Editing vs. CRISPR: Precision in Gene Therapy [https://kactusbio.com/blogs/gene-editing/base-editing-vs-crispr-navigating-precision-gene-therapy?srsltid=AfmBOorHvQZTbqOIABfKhDWLBsAaUStwbPQhu6Z89UzKtc5KeAWTy6u4]
- 60. The comparison of ZFNs, TALENs, and SpCas9 by GUIDE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8655392/]
- 61. CRISPR-Cas9 DNA Base-Editing and Prime-Editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7503568/]
- 62. Back to Basics - Base & Prime Editing [https://frontlinegenomics.com/back-to-basics-base-and-prime-editing/]
- 63. Prime Editing Vs. CRISPR-Cas9 [https://www.fiosgenomics.com/prime-editing-crispr-cas9/]
- 64. CMN Weekly (3 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-3-october-2025-your-weekly-crispr-medicine-news/]
- 65. Advancing genome editing with artificial intelligence [https://pmc.ncbi.nlm.nih.gov/articles/PMC10800897/]
- 66. Improved CRISPR/Cas9 off-target prediction with ... [https://pubmed.ncbi.nlm.nih.gov/41223195/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1-SjSZJSoHBGsnEXcLDjZZ8F-vz5YagNLIUYugSQFZXvZle9Rx&fc=None&ff=20251113024544&v=2.18.0.post22+67771e2]
- 67. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://www.nature.com/articles/s42003-025-08275-6]
- 68. Revolutionizing CRISPR technology with artificial intelligence [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322281/]
- 69. Deep learning predicts CRISPR off-target effects [https://crisprmedicinenews.com/news/deep-learning-predicts-crispr-off-target-effects/]
- 70. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOoraBx9MhZDf1y1dWHtlNEKbWPajvv2epMthQjNhLPT0Pip9nVdR]
- 71. Gene Editing Techniques: ZFNs , TALENs or CRISPR ? [https://www.news-medical.net/life-sciences/Gene-Editing-Techniques-ZFNs-TALENs-or-CRISPR.aspx]
- 72. Improved CRISPR/Cas9 off-target prediction with DNABERT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611124/]
- 73. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144082/]
- 74. High Throughput Screening Market Forecast, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/high-throughput-screening-market]
- 75. 2025 High Throughput Screening (HTS) Industry Trends ... [https://www.openpr.com/news/4125058/2025-high-throughput-screening-hts-industry-trends-report]
- 76. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOoqFPUYXTpsCEn3DnYbd_iZwG2w4J4zw_twL1XVkOEb05E1r95Dm]
- 77. Gene-Editing Could Modify and Cure Disease: CRISPR vs. ... [https://www.ark-invest.com/articles/analyst-research/crispr-vs-talens]
- 78. CRISPR-Cas9, TALENs and ZFNs - the battle in gene editing [https://www.ptglab.com/news/blog/crispr-cas9-talens-and-zfns-the-battle-in-gene-editing/?srsltid=AfmBOopqwyQ0ZwFbx1KIiZCxW9TZoJWJJi-5pW5DZL6xFAOZIjoC2RoP]
- 79. Global Genome Editing Market Size, Trends & Forecast, 2035 [https://www.rootsanalysis.com/reports/genome-editing-market.html]
- 80. Recent applications, future perspectives, and limitations of ... [https://www.sciencedirect.com/science/article/pii/S216225312500188X]