CRISPR Knockout vs RNAi: A 2024 Decision Guide for Functional Genomics Researchers
This comprehensive guide provides researchers, scientists, and drug development professionals with an up-to-date analysis of CRISPR knockout and RNAi technologies for functional genomics.
CRISPR Knockout vs RNAi: A 2024 Decision Guide for Functional Genomics Researchers
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with an up-to-date analysis of CRISPR knockout and RNAi technologies for functional genomics. We explore their foundational principles, delve into detailed methodologies and application-specific workflows, address common troubleshooting and optimization challenges, and present a direct comparative validation framework. The article synthesizes current best practices to empower informed experimental design, enabling the selection of the optimal tool for precise gene function elucidation, target identification, and validation in biomedical research.
Understanding the Core: Foundational Principles of CRISPR Knockout and RNAi
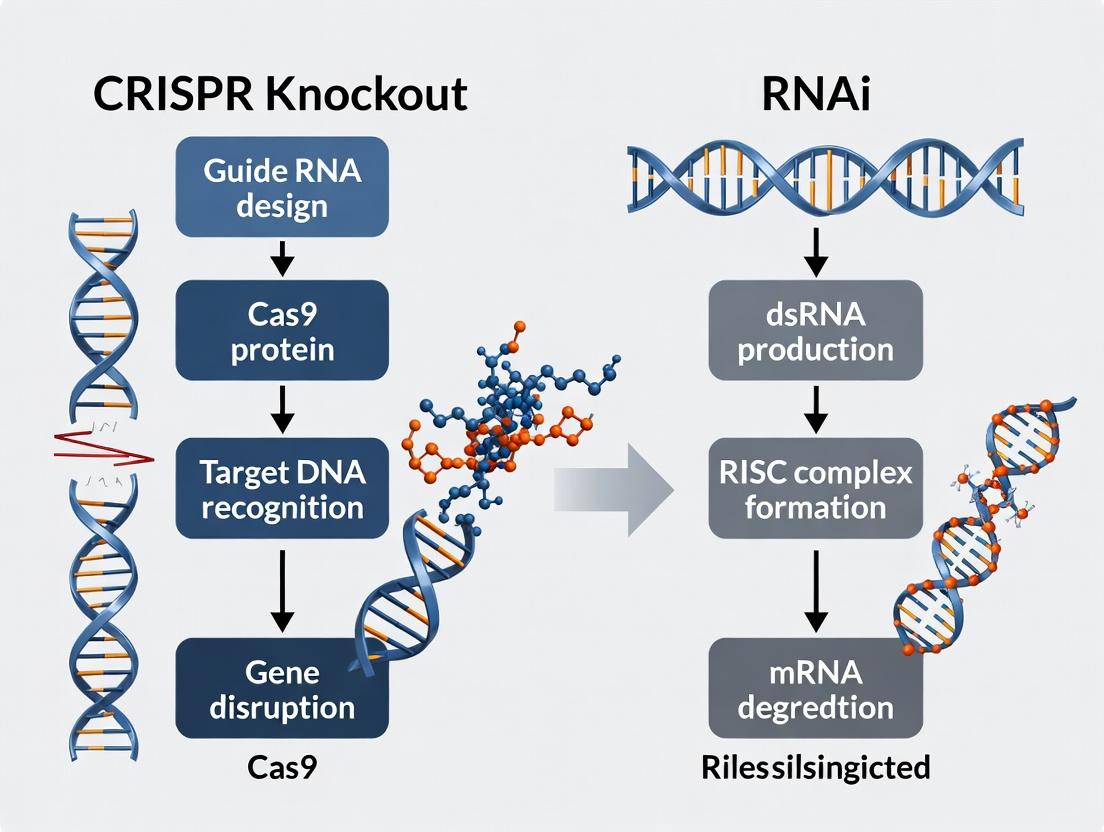
Within the context of functional genomics research, the choice between CRISPR-mediated knockout and RNA interference (RNAi)-mediated knockdown is fundamental. This guide delineates the core mechanisms of permanent DNA editing (exemplified by CRISPR-Cas9) and transient mRNA silencing (exemplified by RNAi), providing a technical framework for researchers and drug development professionals to inform experimental design.
Core Mechanisms and Molecular Pathways
Permanent DNA Editing via CRISPR-Cas9
The CRISPR-Cas9 system creates targeted, heritable double-strand breaks (DSBs) in genomic DNA, which are repaired primarily via Non-Homologous End Joining (NHEJ) or Homology-Directed Repair (HDR). NHEJ is error-prone, leading to insertion/deletion (indel) mutations that can disrupt the open reading frame of a target gene, resulting in a permanent knockout.
Diagram 1: CRISPR-Cas9 Gene Editing Workflow
Transient mRNA Silencing via RNA Interference (RNAi)
RNAi utilizes small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) to guide the RNA-induced silencing complex (RISC) to complementary mRNA sequences. Argonaute 2 (Ago2) within RISC cleaves the target mRNA, leading to its degradation and transient suppression of protein expression without altering the genome.
Diagram 2: RNAi-Mediated Gene Silencing Pathway
Quantitative Comparison of Key Parameters
Table 1: Core Technical & Performance Characteristics
| Parameter | CRISPR-Cas9 Knockout | RNAi (siRNA/shRNA) Knockdown |
|---|---|---|
| Molecular Target | Genomic DNA | mRNA |
| Effect Duration | Permanent, heritable | Transient (days to weeks) |
| Typical Efficiency | 20-80% indel rate (varies by cell type) | 70-95% mRNA reduction (optimized) |
| Off-Target Effects | DNA-level (predicted by guide specificity; lower with high-fidelity Cas9) | RNA-level (seed-based miRNA-like effects; requires careful design) |
| Key Repair Pathway | NHEJ (for knockout) | N/A - Cytoplasmic machinery (RISC) |
| Delivery Common Forms | Plasmid, mRNA, RNP | siRNA (transfection), lentiviral shRNA |
| Phenotype Onset | Slow (requires protein turnover + cell division) | Rapid (hours to days) |
| Primary Use Case | Complete gene ablation, functional nulls, bi-allelic disruption | Acute gene suppression, dose-response studies, essential gene analysis |
Table 2: Experimental Design Considerations for Functional Genomics
| Consideration | CRISPR-Cas9 | RNAi |
|---|---|---|
| Screening Format | Pooled or arrayed (requires cloning/validation of guides) | Pooled (lentiviral shRNA) or arrayed (siRNA/siRNA libraries) |
| Optimal Readout Time | ≥ 5-7 days post-editing (allows for genomic fixation) | 48-96 hours post-transfection (peak knockdown) |
| Control Requirements | Non-targeting guide, mock-treated, Cas9-only | Non-targeting siRNA, scramble shRNA, transfection reagent |
| Validation Necessity | Essential: Sequencing of target locus, protein null confirmation | Essential: qPCR for mRNA, Western blot for protein |
| Cost & Throughput | Higher upfront cost (cloning, sequencing); high-throughput possible | Generally lower cost per sample; mature high-throughput protocols |
Detailed Methodological Protocols
Protocol for CRISPR-Cas9 Knockout Validation
Aim: To generate and validate a clonal knockout cell line. Key Steps:
- sgRNA Design & Cloning: Design two sgRNAs targeting early exons of the gene of interest (GOI). Clone into a CRISPR plasmid (e.g., lentiCRISPRv2) expressing both sgRNA and SpCas9.
- Delivery & Transduction: Package lentivirus and transduce target cells at low MOI. Apply appropriate selection (e.g., puromycin) for 3-5 days.
- Single-Cell Cloning: Serial dilute polyclonal population to 0.5 cells/well in a 96-well plate. Expand clones for 2-3 weeks.
- Genomic DNA Extraction: Harvest cells from each clone. Use a kit (e.g., QuickExtract) for rapid gDNA isolation.
- PCR & Sequencing: PCR amplify the target region (~500-700 bp flanking cut site). Purify PCR product and perform Sanger sequencing.
- Sequence Analysis: Use tools like TIDE (Tracking of Indels by DEcomposition) or ICE (Inference of CRISPR Edits) to quantify editing efficiency and infer indel sequences from chromatogram data. For clonal lines, sequence alignment to reference is required to confirm bi-allelic disruption.
- Phenotypic Validation: Perform Western blot to confirm loss of target protein.
Protocol for RNAi Knockdown Validation
Aim: To achieve and validate acute gene knockdown. Key Steps:
- siRNA Design & Selection: Use validated siRNA sequences from public databases (e.g., Dharmacon, Sigma) or design using algorithms. Always use a pool of 3-4 siRNAs per gene to mitigate off-targets.
- Reverse Transfection: Plate cells in antibiotic-free medium. Complex siRNA (10-50 nM final) with lipid-based transfection reagent (e.g., Lipofectamine RNAiMAX) in Opti-MEM. Add complex to cells.
- Optimal Harvest Time: Harvest cells for RNA analysis at 48 hours post-transfection. Harvest for protein analysis at 72-96 hours.
- mRNA-Level Validation (qRT-PCR): Isolate total RNA, reverse transcribe to cDNA. Perform quantitative PCR using TaqMan or SYBR Green assays with primers for the GOI. Normalize to housekeeping genes (e.g., GAPDH, ACTB). Calculate fold-change using the ΔΔCt method.
- Protein-Level Validation (Western Blot): Lyse cells in RIPA buffer. Separate proteins by SDS-PAGE, transfer to membrane, and probe with antibodies against target protein and a loading control (e.g., β-actin). Densitometry quantifies knockdown efficiency.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR and RNAi Experiments
| Reagent Category | Specific Example(s) | Function in Experiment |
|---|---|---|
| CRISPR-Cas9 Systems | lentiCRISPRv2 plasmid, Alt-R S.p. Cas9 Nuclease V3, synthetic sgRNA | Provides the enzymatic machinery and targeting guide for DNA cleavage. High-fidelity Cas9 variants reduce off-targets. |
| RNAi Triggers | ON-TARGETplus siRNA SMARTpools, MISSION shRNA Lentiviral Particles | Induces sequence-specific degradation of target mRNA. Pools mitigate off-target effects. |
| Delivery Vehicles | Lipofectamine CRISPRMAX, Lipofectamine RNAiMAX, Polybrene (for viral transduction) | Enables efficient intracellular delivery of nucleic acids or RNPs. Critical for hard-to-transfect cells. |
| Editing Detection | T7 Endonuclease I, Alt-R Genome Editing Detection Kit, Sanger Sequencing | Detects and quantifies the presence of indels at the target genomic locus. |
| Knockdown Validation | TaqMan Gene Expression Assays, SYBR Green Master Mix, Target-specific antibodies (for Western) | Quantifies reduction in target mRNA or protein levels to confirm knockdown efficacy. |
| Selection & Cloning | Puromycin, Blasticidin, Limiting Dilution Cloning Tools | Selects for successfully transduced cells and enables isolation of single-cell clones (CRISPR). |
| Cell Culture Media | Opti-MEM Reduced Serum Media | Used during transfection complex formation to reduce toxicity and enhance efficiency. |
Historical Context and Technological Evolution in Functional Genomics
Functional genomics seeks to understand the complex relationship between genotype and phenotype. Its history is marked by pivotal technological shifts, each enabling more precise and systematic interrogation of gene function. This evolution is critically framed by the ongoing methodological debate: the use of RNA interference (RNAi) for gene knockdown versus CRISPR-Cas9 for complete gene knockout. This guide details the historical progression, current protocols, and quantitative comparisons central to this discourse.
Historical Timeline and Technological Milestones
The field originated with forward genetics, moving to targeted reverse genetics with the advent of RNAi, and finally to programmable nuclease systems.
Table 1: Key Technological Milestones
| Era | Technology (Year) | Core Principle | Primary Application in Functional Genomics | Major Limitation |
|---|---|---|---|---|
| Pre-1990s | Chemical & Radiation Mutagenesis | Random induction of mutations | Forward genetic screens | Low throughput, arduous gene identification |
| 1998 | RNA Interference (RNAi) | dsRNA-triggered post-transcriptional mRNA degradation | Targeted gene knockdown in mammalian cells | Off-target effects, incomplete knockdown |
| 2006 | siRNA Libraries | Synthetic siRNA pools | High-throughput loss-of-function screens | Transient effect, high false-positive rates |
| 2009 | shRNA Libraries | Viral delivery of short-hairpin RNAs | Stable gene knockdown screens | Still incomplete knockdown, miRNA-like off-targets |
| 2013 | CRISPR-Cas9 Knockout | Cas9 nuclease creates double-strand breaks, repaired by error-prone NHEJ | Permanent, complete gene knockout | Homology-directed repair can confound, editing efficiency varies |
| 2015-2017 | CRISPRi/a | Catalytically dead Cas9 (dCas9) fused to repressors/activators | Reversible knockdown or transcriptional activation | More consistent than RNAi, but still a knockdown (CRISPRi) |
Core Experimental Methodologies
Genome-Scale RNAi Screening Protocol
This protocol uses lentiviral shRNA libraries for stable integration.
Key Steps:
- Library Design & Cloning: A pooled lentiviral library is constructed, typically containing 3-6 shRNAs per gene, plus non-targeting controls.
- Virus Production: HEK293T cells are transfected with the shRNA library plasmid, packaging (psPAX2), and envelope (pMD2.G) plasmids.
- Cell Infection & Selection: Target cells are infected at a low MOI (<0.3) to ensure single integration. Puromycin selection is applied for 48-72 hours.
- Screen Execution: The selected cell population is divided: one portion harvested as the "T0" reference, the other subjected to the selective pressure (e.g., drug treatment, growth over time).
- Genomic DNA Extraction & Sequencing: Genomic DNA is isolated from T0 and endpoint populations. The integrated shRNA barcodes are PCR-amplified and deep-sequenced.
- Data Analysis: shRNA abundance changes between T0 and endpoint are calculated. Gene-level scores are derived by robust statistical aggregation (e.g., RIGER, RSA) of multiple shRNAs per gene.
CRISPR-Cas9 Knockout Screening Protocol
This protocol utilizes a single-guide RNA (sgRNA) library to direct Cas9-mediated gene knockout.
Key Steps:
- Cell Line Engineering: A target cell line is engineered to stably express Cas9 nuclease.
- sgRNA Library Design & Cloning: A pooled lentiviral sgRNA library is used, typically with 3-10 guides per gene, targeting early exons. Non-targeting and essential gene-targeting controls are included.
- Viral Transduction & Selection: Cas9-expressing cells are transduced at low MOI (<0.3) and selected with puromycin.
- Screen Execution & Sample Collection: As with RNAi, cells are collected at T0 and after selection. For negative selection (e.g., cell fitness), the endpoint is after ~14 population doublings.
- Next-Generation Sequencing (NGS) Prep: Genomic DNA is extracted. The sgRNA sequences are amplified with primers containing Illumina adapters and sample barcodes.
- Analysis: Read counts per sgRNA are normalized. Gene essentiality scores (e.g., MAGeCK, CERES) are computed, correcting for copy number effects and multiple guides per gene.
Table 2: Quantitative Comparison of RNAi vs. CRISPR-KO Screening Data
| Parameter | RNAi (shRNA) Screening | CRISPR-KO Screening |
|---|---|---|
| Typical Effect on Target | Transcriptional Knockdown (70-90% reduction) | Complete Gene Disruption (Frameshift mutations) |
| On-target Efficacy Variance | High (due to variable shRNA processing) | Generally High and More Consistent |
| False Negative Rate | Higher (incomplete knockdown can mask phenotype) | Lower (complete knockout reveals full effect) |
| False Positive Rate (Off-target) | Significant (miRNA-like seed region effects) | Lower (requires >17-nt seed match; can use improved nucleases like SpCas9-HF1) |
| Typical Screen Consistency (Pearson R between replicates) | 0.7 - 0.85 | 0.9 - 0.98 |
| Optimal Screen Duration | Shorter-term assays (days to 1-2 weeks) | Longer-term viable (weeks) due to permanent edit |
| Key Analytical Metric | Gene-level RIGER score, Z-score | MAGeCK beta score, CERES probability |
Visualization of Core Concepts
Title: RNAi vs CRISPR-Cas9 Gene Inactivation Pathways
Title: Functional Genomics Pooled Screening Workflow
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Research Reagents for Functional Genomics Screens
| Reagent/Material | Function & Description | Example Vendor/Product |
|---|---|---|
| Genome-wide shRNA Library | Pooled lentiviral plasmids encoding short-hairpin RNAs for targeted mRNA knockdown. | Horizon (Dharmacon) GIPZ or TRIPZ libraries; Sigma MISSION shRNA library. |
| Genome-wide CRISPR Knockout (sgRNA) Library | Pooled lentiviral plasmids encoding single-guide RNAs targeting genes for Cas9-mediated knockout. | Broad Institute Brunello or Dolcetto libraries; Addgene library collections. |
| Lentiviral Packaging Mix | Plasmid mix (e.g., psPAX2, pMD2.G) for producing replication-incompetent lentivirus in HEK293T cells. | Invitrogen Virapower Mix; Addgene packaging plasmids. |
| Polybrene (Hexadimethrine Bromide) | A cationic polymer that enhances viral transduction efficiency by neutralizing charge repulsion. | Sigma-Aldrich H9268. |
| Puromycin Dihydrochloride | Antibiotic for selecting cells successfully transduced with shRNA or sgRNA vectors containing a puromycin resistance gene. | Thermo Fisher Scientific A1113803. |
| Next-Generation Sequencing Kit | For preparing NGS libraries from amplified sgRNA or shRNA barcodes. | Illumina Nextera XT; NEBNext Ultra II DNA. |
| Cas9-Stable Cell Line | A cell line constitutively expressing the Cas9 nuclease, required for CRISPR-KO screens. | Generated in-house or available from ATCC (e.g., HEK293-Cas9). |
| Cell Viability/Proliferation Assay | To measure screen phenotype (e.g., CellTiter-Glo for ATP-based viability). | Promega CellTiter-Glo Luminescent Assay. |
| Genomic DNA Extraction Kit | For high-yield, high-purity gDNA from screen cell pellets for NGS library prep. | Qiagen Blood & Cell Culture DNA Maxi Kit. |
Within functional genomics research, two principal technologies dominate loss-of-function studies: CRISPR-Cas9-mediated knockout and RNA interference (RNAi)-mediated knockdown. The efficacy, specificity, and durability of each approach are dictated by their distinct underlying molecular machineries. This whitepaper provides an in-depth technical comparison of the core molecular players—the Cas9/gRNA complex for CRISPR and the siRNA/shRNA/Dicer/RISC pathway for RNAi—framed within the critical decision-making context of functional genomics and drug target validation.
The RNAi Pathway: siRNA/shRNA, Dicer, and RISC
RNAi is a conserved eukaryotic pathway for sequence-specific post-transcriptional gene silencing. It utilizes endogenous cellular machinery to degrade target messenger RNA (mRNA).
Core Molecular Players
- siRNA (Small Interfering RNA): Typically 21-23 bp double-stranded RNA (dsRNA) duplexes with 2-nt 3' overhangs. Synthetic siRNAs are directly transfected into cells.
- shRNA (Short Hairpin RNA): DNA-encoded RNA molecules with a tight hairpin turn. Delivered via viral vectors, they are transcribed in the nucleus and exported to the cytoplasm.
- Dicer: A cytoplasmic endoribonuclease (RNase III family) that processes long dsRNA or shRNA into mature siRNA duplexes.
- RISC (RNA-Induced Silencing Complex): A multi-protein complex anchored by an Argonaute (Ago2) family protein. RISC loading involves unwinding of the siRNA duplex, retention of the guide strand, and degradation of the passenger strand. The guide strand then directs RISC to complementary mRNA targets.
- Ago2 (Argonaute 2): The catalytic engine of RISC, responsible for "slicing" (cleaving) the target mRNA between nucleotides complementary to positions 10 and 11 of the guide strand.
Mechanism & Pathway
The pathway for shRNA-mediated silencing is depicted below.
Diagram Title: RNAi Pathway: shRNA Processing and RISC Action
Key Experimental Protocol: shRNA-Mediated Knockdown
Objective: Achieve stable, long-term gene knockdown in a mammalian cell line. Workflow:
- Design & Cloning: Design 19-21 nt target sequences meeting specificity and thermodynamic guidelines. Clone oligonucleotides into an shRNA expression plasmid (e.g., pLKO.1) downstream of a U6 or H1 Pol III promoter.
- Virus Production: Co-transfect the shRNA plasmid with packaging plasmids (psPAX2, pMD2.G) into HEK293T cells using PEI or calcium phosphate. Harvest lentiviral supernatant at 48 and 72 hours.
- Cell Transduction: Incubate target cells with viral supernatant plus polybrene (8 µg/mL). Spinoculation (centrifugation at 600-1000 x g for 30-90 min at 32°C) enhances efficiency.
- Selection & Validation: Apply selection antibiotic (e.g., Puromycin, 1-5 µg/mL) 48 hours post-transduction for 3-7 days. Validate knockdown via qRT-PCR (mRNA) and western blot (protein) 5-7 days post-selection.
The CRISPR-Cas9 Pathway: Cas9 and gRNA
CRISPR-Cas9 is a programmable DNA endonuclease system derived from bacterial adaptive immunity, enabling permanent genomic modification.
Core Molecular Players
- Cas9 (CRISPR-associated protein 9): A DNA endonuclease that generates double-strand breaks (DSBs). The commonly used Streptococcus pyogenes Cas9 (SpCas9) recognizes the PAM sequence 5'-NGG-3'.
- gRNA (guide RNA): A chimeric RNA composed of a ~20 nt CRISPR RNA (crRNA) that specifies the target DNA sequence and a trans-activating crRNA (tracrRNA) scaffold that binds Cas9. Often expressed as a single guide RNA (sgRNA).
- PAM (Protospacer Adjacent Motif): A short, fixed sequence adjacent to the target DNA that is essential for Cas9 recognition and binding. Not present in the host's CRISPR locus.
Mechanism & Pathway
The pathway for CRISPR-Cas9 mediated DNA cleavage and knockout is depicted below.
Diagram Title: CRISPR-Cas9 Pathway: DNA Cleavage and Knockout
Key Experimental Protocol: CRISPR-Cas9 Knockout via NHEJ
Objective: Generate a homozygous, frameshift knockout in a diploid mammalian cell line. Workflow:
- gRNA Design & Validation: Select 20-nt target sequences immediately 5' to an NGG PAM using validated algorithms (e.g., from the Broad Institute). Prioritize on-target efficiency scores and minimize off-target potential. Validate cutting efficiency via T7 Endonuclease I (T7E1) or ICE assay before stable line generation.
- Delivery: For high efficiency, use electroporation of pre-assembled Cas9-gRNA RNP complexes. Alternatively, co-transfect a Cas9 expression plasmid (or mRNA) and a gRNA expression plasmid.
- Clonal Isolation: 48-72 hours post-delivery, single-cell sort or perform limiting dilution in 96-well plates. Expand clones for 2-3 weeks.
- Genotyping & Validation:
- Extract genomic DNA from clones.
- PCR-amplify the target locus (amplicon: 400-600 bp).
- Sequence the PCR product by Sanger sequencing.
- Analyze chromatograms for overlapping peaks downstream of the cut site. Use decomposition software (e.g., ICE, TIDE) to calculate indel efficiency or identify biallelic frameshift mutations.
- Confirm protein loss via western blot.
Quantitative Comparison of Core Properties
Table 1: Molecular & Mechanistic Comparison
| Property | CRISPR-Cas9 System | RNAi (siRNA/shRNA) Pathway |
|---|---|---|
| Target Molecule | Genomic DNA | Cytoplasmic mRNA |
| Primary Effect | Permanent DNA cleavage | mRNA degradation / translational inhibition |
| Key Enzymes | Cas9 endonuclease | Dicer (processor), Argonaute 2 (slicer) |
| Guide Component | ~100 nt gRNA (crRNA+tracrRNA) | ~21-23 nt siRNA guide strand |
| Sequence Recognition | DNA-DNA complementarity + PAM | RNA-RNA complementarity (seed region: nt 2-8) |
| Typical Efficiency | High (often >70% indel formation) | Variable (0-90% knockdown) |
| Typical Off-Target Effects | DNA-level mismatches, especially distal from PAM | miRNA-like seed region off-targets |
| Duration of Effect | Permanent, heritable | Transient (siRNA: days) / stable (shRNA: weeks) |
Table 2: Functional Genomics Application Context
| Consideration | CRISPR Knockout | RNAi Knockdown |
|---|---|---|
| Goal | Complete loss-of-function, study of essential genes, create mutant models | Partial reduction (hypomorph), study dose-sensitive genes, rapid screening |
| Best For | Phenotypes from complete protein ablation; genes with long protein half-life | Phenotypes sensitive to protein dosage; essential gene analysis; acute inhibition |
| Key Limitation | Clonal variability; genomic context (PAM requirement); potential for compensatory mutations | Residual protein activity; incomplete knockdown; potential for saturation & competition of endogenous miRNA |
| Throughput | High (arrayed or pooled screens) | Very High (arrayed or pooled screens) |
| Druggability Insight | Mimics complete inhibitor blockade | Mimics partial inhibitor effect |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Core Experiments
| Reagent / Material | Function & Explanation | Typical Example (Vendor) |
|---|---|---|
| pLKO.1-puro Vector | Lentiviral shRNA expression backbone. Contains Puromycin resistance for stable selection. | TRC Lentiviral shRNA plasmids (Dharmacon) |
| Lentiviral Packaging Mix | Plasmid mix (psPAX2, pMD2.G) providing gag/pol and VSV-G envelope proteins for virus production. | MISSION Lentiviral Packaging Mix (Sigma-Aldrich) |
| Lipofectamine RNAiMAX | Cationic lipid reagent optimized for high-efficiency, low-cytotoxicity delivery of siRNA. | Lipofectamine RNAiMAX (Thermo Fisher) |
| pSpCas9(BB)-2A-Puro (PX459) | All-in-one plasmid expressing SpCas9, a gRNA scaffold, and Puromycin resistance. Streamlines knockout generation. | pX459 V2.0 (Addgene #62988) |
| Alt-R S.p. Cas9 Nuclease V3 | High-purity, recombinant Cas9 protein for RNP formation. Enables rapid, DNA-free delivery with reduced off-target activity. | Alt-R S.p. Cas9 Nuclease (IDT) |
| T7 Endonuclease I | Surveyor nuclease. Detects mismatches in heteroduplex DNA formed by annealing WT and mutant PCR products, quantifying indel efficiency. | T7 Endonuclease I (NEB) |
| Polybrene | Cationic polymer that neutralizes charge repulsion between viral particles and cell membrane, enhancing transduction efficiency. | Hexadimethrine bromide (Sigma-Aldrich) |
| ClonaCell-TCS Medium | Semi-solid methylcellulose medium for isolating single mammalian cell colonies post-transfection/transduction. | ClonaCell (STEMCELL Technologies) |
Within functional genomics and drug target validation, a fundamental strategic decision is whether to pursue complete gene ablation (e.g., via CRISPR-Cas9 knockout) or transcript knockdown (e.g., via RNAi). This choice is not trivial; it dictates experimental outcomes, biological interpretations, and therapeutic insights. This guide provides a technical framework for making this decision, situated within the broader thesis that CRISPR knockout and RNAi are complementary technologies, each with distinct applications defined by the primary research objective.
Core Technology Comparison: Mechanisms & Outcomes
Fundamental Mechanisms
- CRISPR-Cas9 Knockout: Utilizes a Cas9 endonuclease guided by a single-guide RNA (sgRNA) to create a double-strand break (DSB) at a specific genomic locus. Repair via error-prone non-homologous end joining (NHEJ) often results in insertions or deletions (indels) that disrupt the open reading frame, leading to permanent, complete ablation of the gene and its protein product.
- RNA Interference (RNAi): Employs exogenous small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) that are loaded into the RNA-induced silencing complex (RISC). RISC uses the guide strand to bind complementary mRNA sequences, leading to transcript cleavage (siRNA) or translational repression and transcript degradation (shRNA). This results in transient, partial knockdown of gene expression.
Quantitative Performance Metrics
The table below summarizes key performance differences, critical for experimental planning.
Table 1: Core Characteristics of Gene Ablation vs. Transcript Knockdown
| Parameter | CRISPR-Cas9 Knockout | RNAi (siRNA/shRNA) |
|---|---|---|
| Target | Genomic DNA | mRNA transcript |
| Modification | Permanent, heritable | Transient (siRNA) or stable (shRNA) |
| Efficacy (Protein Reduction) | Often 100% (complete null) | Typically 70-95% (knockdown) |
| Onset of Effect | Slow (requires protein turnover) | Rapid (hours to days) |
| Duration of Effect | Permanent in cell line | 3-7 days (siRNA); can be stable (shRNA) |
| Primary Pitfalls | Off-target indels, clone heterogeneity | Off-target transcript effects, incomplete knockdown, miRNA-like effects |
| Optimal Use Case | Essential gene analysis, study of non-coding regions, long-term phenotype | Dose-response studies, essential gene titration, acute inhibition, in vivo delivery |
Strategic Decision Framework: When to Choose Which
The primary research objective is the paramount deciding factor.
Aim for Complete Gene Ablation (CRISPR Knockout) When:
- Studying Essential Genes: To generate hypomorphic or null clones that survive via adaptation or compensator mechanisms, revealing core gene function.
- Eliminating All Protein Isoforms: When the target gene has multiple splice variants, and the objective is to disrupt all potential protein products.
- Investigating Non-Coding Genomic Regions: For functional screens of enhancers, promoters, or long non-coding RNA genes.
- Requiring a Clean Genetic Background: For studies where even low residual protein expression could confound results (e.g., signaling pathway on/off states).
- Long-Term Phenotypic Studies: Such as cellular differentiation, senescence, or long-term in vivo xenograft models.
Aim for Transcript Knockdown (RNAi) When:
- Modeling Therapeutic Inhibition: Most drug candidates are inhibitors, not ablators. RNAi's partial knockdown better mimics pharmacologic inhibition.
- Titrating Gene Dosage: To study haploinsufficiency or establish a correlation between expression level and phenotypic severity.
- Studying Acute Phenotypes in Essential Genes: Where permanent knockout would be lethal, allowing observation of immediate consequences before compensatory mechanisms arise.
- Working with Difficult-to-Edit Cells: Such as primary cells or post-mitotic cells where HDR/NHEJ efficiency is low.
- Rapid, High-Throughput Screening: siRNA libraries remain a robust, cost-effective tool for initial target identification.
Title: Decision Framework for Gene Ablation vs. Knockdown
Detailed Experimental Protocols
Protocol for CRISPR-Cas9 Knockout Validation
This protocol ensures confirmation of bi-allelic frameshift mutations.
Key Steps:
- sgRNA Design & Cloning: Design two sgRNAs targeting early exons. Clone into a Cas9-expression plasmid (e.g., lentiCRISPRv2).
- Delivery & Selection: Transduce target cells. Apply appropriate selection (e.g., puromycin) for 5-7 days.
- Single-Cell Cloning: Dilute cells to ~0.5 cells/well in a 96-well plate. Expand clones for 2-3 weeks.
- Genomic DNA Extraction: Use a commercial kit to extract gDNA from each clone.
- PCR & Sanger Sequencing: PCR amplify the target region (approx. 500-700bp). Submit for Sanger sequencing.
- Sequence Analysis: Use chromatogram decomposition tools (e.g., ICE Analysis, Synthego) to quantify editing efficiency and infer indel sequences. Confirm bi-allelic frameshifts.
- Western Blot Validation: Perform immunoblot to confirm complete absence of target protein.
Protocol for Optimized RNAi Knockdown
This protocol minimizes off-target effects and confirms knockdown efficacy.
Key Steps:
- siRNA Design/Pooling: Use a pool of 4-5 distinct siRNAs targeting different regions of the transcript, or validated shRNA constructs.
- Reverse Transfection: For siRNA, use a lipid-based transfection reagent. Seed cells directly into siRNA-lipid complexes for high efficiency.
- Dose & Time Optimization: Titrate siRNA concentration (e.g., 1-50 nM) and harvest cells at 48, 72, and 96 hours post-transfection.
- qRT-PCR Validation: Isolate RNA, synthesize cDNA, and perform qPCR using TaqMan assays to measure transcript depletion (optimal: >80%).
- Protein-Level Validation: Perform Western blot at 72-96 hours to confirm protein reduction.
- Rescue Experiment: Express an siRNA-resistant cDNA version of the target gene to confirm phenotype specificity.
Title: CRISPR vs RNAi Experimental Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Gene Perturbation Studies
| Reagent / Solution | Function & Description | Example (Non-exhaustive) |
|---|---|---|
| CRISPR-Cas9 All-in-One Vector | Combines expression of Cas9, sgRNA, and a selection marker (e.g., puromycin) for stable knockout generation. | lentiCRISPRv2, pSpCas9(BB)-2A-Puro |
| High-Fidelity Cas9 Variant | Reduces off-target DNA cleavage while maintaining on-target efficacy. | SpCas9-HF1, eSpCas9(1.1) |
| Sanger Sequencing Analysis Tool | Software to deconvolute complex chromatograms from edited polyclonal or early-clonal populations. | ICE (Synthego), TIDE, Tracking of Indels by DEcomposition |
| Validated siRNA SMARTpools | Pre-designed, pooled siRNAs targeting a single gene with multiple sequences to enhance potency and reduce off-targets. | Dharmacon ON-TARGETplus, Qiagen FlexiTube |
| Lipid-Based Transfection Reagent | For high-efficiency, low-toxicity delivery of siRNA into mammalian cells. | Lipofectamine RNAiMAX, DharmaFECT |
| shRNA Lentiviral Particles | For stable, long-term knockdown; delivered via viral transduction. | Mission TRC shRNAs (Sigma), GIPZ shRNAs (Horizon) |
| cDNA Rescue Construct | Expression vector containing the target gene cDNA with silent mutations in the siRNA target site to confirm phenotype specificity. | Custom gene synthesis and cloning into pcDNA3.1 or pLX307. |
Integrated Analysis & Validation
Regardless of the method chosen, rigorous validation is non-negotiable.
- Phenotypic Correlation: The strongest phenotype from a complete knockout may not be the most therapeutically relevant. Compare knockout and knockdown phenotypes side-by-side.
- Multi-Method Convergence: Confidence in a gene's role increases when congruent phenotypes are observed with both CRISPR knockout and RNAi knockdown (using distinct reagents).
- Off-Target Assessment: For CRISPR, use GUIDE-seq or orthogonal Sanger sequencing of predicted off-target sites. For RNAi, always include multiple siRNA sequences targeting the same gene and a rescue experiment.
The decision between aiming for complete gene ablation or transcript knockdown is foundational. CRISPR knockout provides a definitive, permanent genetic null, ideal for understanding fundamental biology and the function of all gene products. RNAi knockdown offers a transient, titratable reduction that more closely mirrors pharmacological intervention and allows study of essential genes. The informed researcher selects the tool based on the biological question, often employing both in a complementary strategy to build robust, clinically relevant insights in functional genomics and drug discovery.
Inherent Strengths and Limitations of Each Platform from First Principles
Functional genomics aims to understand gene function and interaction, with gene perturbation being a cornerstone methodology. Two primary platforms dominate this field: RNA interference (RNAi) and CRISPR-Cas9 mediated knockout. Framed within the broader thesis of selecting the optimal tool for functional genomics research, this guide analyzes each platform from first principles—examining their foundational biological mechanisms to derive their inherent operational strengths and limitations. The choice between these platforms significantly impacts the interpretation of data in target identification, validation, and drug development.
First Principles: Core Biological Mechanisms
RNA Interference (RNAi): RNAi is a conserved eukaryotic post-transcriptional gene silencing pathway. Exogenous introduction of small interfering RNA (siRNA) or short hairpin RNA (shRNA) leads to loading of the RNA-induced silencing complex (RISC). The guide strand directs RISC to complementary mRNA sequences, resulting in Argonaute-catalyzed cleavage or translational inhibition of the target mRNA. Protein levels are reduced, but the genomic DNA remains unaltered.
CRISPR-Cas9 Knockout: The CRISPR-Cas9 system, adapted from a prokaryotic immune defense, creates permanent genomic alterations. A single guide RNA (sgRNA) directs the Cas9 nuclease to a specific genomic locus complementary to a 20-nucleotide sequence adjacent to a Protospacer Adjacent Motif (PAM). Cas9 induces a double-strand break (DSB), which is repaired by error-prone Non-Homologous End Joining (NHEJ), often resulting in insertions or deletions (indels) that disrupt the open reading frame, leading to a complete loss of function.
The following tables synthesize key performance metrics for both platforms, derived from current literature and experimental benchmarks.
Table 1: Fundamental Characteristics & Performance Metrics
| Parameter | RNAi (si/shRNA) | CRISPR-Cas9 Knockout | Notes |
|---|---|---|---|
| Primary Target | mRNA | Genomic DNA | Fundamental difference in mechanism. |
| Effect on Gene | Transcript knockdown (reduction) | Gene knockout (disruption) | RNAi is typically incomplete; CRISPR aims for null alleles. |
| Onset of Effect | Hours to days | Days to weeks | RNAi acts on existing mRNA; CRISPR requires cell division for NHEJ repair and protein turnover. |
| Duration of Effect | Transient (days to a week) | Permanent & heritable | shRNA can be longer-lasting via viral integration. |
| Typical Knockdown Efficiency | 70-95% | Often >95% (frameshift indels) | RNAi efficiency varies by target site and reagent. CRISPR efficiency depends on sgRNA design and delivery. |
| Multiplexing Capacity | High (pools of siRNAs) | High (multiple sgRNAs) | Both enable genome-wide screens. CRISPR libraries require careful design to avoid cross-guide interactions. |
| Key Technical Risk | Off-target effects (seed-sequence mediated) | Off-target effects (sgRNA homology at distal sites) | RNAi off-targets are transcriptome-wide; CRISPR off-targets are genome-wide but less frequent with high-fidelity Cas9 variants. |
| Phenotype Confounding | Cytotoxicity, immune activation (e.g., interferon response) | p53-mediated DNA damage response, copy number alterations | Controls (e.g., non-targeting guides, rescue experiments) are critical. |
Table 2: Suitability for Key Research Applications
| Application | Recommended Platform | Rationale & Considerations |
|---|---|---|
| Acute Gene Inhibition | RNAi | Faster onset, suitable for studying essential genes in short-term assays. |
| Studies of Essential Genes | CRISPR Knockout (in inducible systems) | Conditional knockout avoids cell death during establishment. RNAi can be used for acute depletion. |
| Generating Stable Cell Lines | CRISPR-Cas9 | Creates permanent, defined genetic modifications for consistent study. |
| Genome-Wide Loss-of-Function Screens | Both, with different outputs | RNAi: Hypomorphs, can reveal dose-sensitive phenotypes. CRISPR: Complete knockouts, fewer false negatives, identifies essential genes more robustly. |
| Studying Multigene Families / Redundancy | CRISPR-Cas9 | Ability to target highly homologous genomic regions with precise sgRNA design; RNAi may cross-react. |
| In vivo Models (rodent) | CRISPR-Cas9 | Enables direct, rapid generation of knockout models via embryo injection. RNAi requires sustained delivery. |
Detailed Experimental Protocols
Protocol 1: Genome-Wide CRISPR Knockout Screen (Lentiviral Pooled Screen) Objective: To identify genes essential for cell proliferation under a specific condition (e.g., drug treatment).
- Library Selection: Choose a genome-wide sgRNA library (e.g., Brunello, Brie). The library is cloned into a lentiviral vector containing the sgRNA scaffold and a selectable marker (e.g., puromycin resistance).
- Virus Production: Produce lentiviral particles of the sgRNA library at a low MOI (<0.3) in HEK293T cells using standard packaging plasmids (psPAX2, pMD2.G).
- Cell Infection & Selection: Infect the target cell line (e.g., a cancer cell line) with the lentiviral library pool. Ensure >200x library coverage (e.g., 50 million cells for a 75,000-guide library). Select transduced cells with puromycin (e.g., 2 µg/mL for 5-7 days).
- Screen Execution: Split the selected cell population into experimental (e.g., treated with drug) and control (DMSO) arms. Maintain cultures for ~14-21 population doublings, keeping >500x library coverage at all times.
- Genomic DNA Extraction & Sequencing: Harvest cells at endpoint. Extract genomic DNA (gDNA) using a large-scale kit (e.g., Qiagen Blood & Cell Culture DNA Maxi Kit). PCR amplify the integrated sgRNA sequences from the gDNA using indexing primers for NGS.
- Data Analysis: Sequence PCR amplicons on an Illumina platform. Align reads to the reference library. Use specialized algorithms (e.g., MAGeCK, CERES) to compare sgRNA abundance between control and experimental arms, identifying significantly depleted or enriched guides/genes.
Protocol 2: Targeted RNAi Knockdown Validation Assay Objective: To validate a hit from a screen or hypothesis by targeted mRNA knockdown.
- siRNA Design & Transfection: Select 2-3 validated siRNAs targeting different regions of the gene of interest. Include a non-targeting control (NTC) siRNA and a positive control (e.g., siRNA against a housekeeping gene). Reverse-transfect cells in a 96-well plate using a lipid-based transfection reagent (e.g., Lipofectamine RNAiMAX) according to manufacturer's protocol (typical siRNA concentration: 10-25 nM).
- Efficiency Validation (qRT-PCR): 48 hours post-transfection, lyse cells for RNA extraction (e.g., using TRIzol). Synthesize cDNA. Perform quantitative PCR (qPCR) using TaqMan or SYBR Green assays specific for the target gene. Normalize Ct values to a housekeeping gene (e.g., GAPDH) and calculate % knockdown relative to NTC using the 2^(-ΔΔCt) method.
- Phenotypic Assessment: In parallel, perform the relevant phenotypic assay (e.g., cell viability assay via ATP quantitation, immunoblotting for pathway proteins, or flow cytometry) at 72-96 hours post-transfection.
- Data Correlation: Correlate the degree of mRNA knockdown (from qPCR) with the magnitude of the phenotypic effect. Concordance across multiple independent siRNAs strengthens the conclusion that the phenotype is on-target.
Visualizations
Diagram Title: RNAi Mechanism: Post-Transcriptional Silencing
Diagram Title: CRISPR-Cas9 Mechanism: Genomic Knockout
Diagram Title: Platform Selection Decision Logic
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for CRISPR and RNAi Experiments
| Reagent / Material | Function in Experiment | Typical Example / Note |
|---|---|---|
| Validated siRNA Pools | Provide consistent, potent knockdown of a specific target with reduced off-target effects compared to single siRNAs. | ON-TARGETplus (Horizon), Silencer Select (Thermo Fisher). |
| Genome-Wide sgRNA Library | Collection of lentiviral vectors encoding sgRNAs targeting every gene in the genome for large-scale screens. | Brunello (human) or Mouse Brie library (Broad Institute). |
| High-Fidelity Cas9 Nuclease | Engineered Cas9 variant (e.g., SpCas9-HF1, eSpCas9) with reduced off-target DNA cleavage activity. | Critical for experiments requiring high specificity, such as therapeutic target validation. |
| Lipid-Based Transfection Reagent (RNAi) | Forms complexes with siRNA to facilitate cellular uptake and endosomal escape into the cytoplasm. | Lipofectamine RNAiMAX (Thermo Fisher). |
| Lentiviral Packaging Plasmids | Required to produce replication-incompetent lentiviral particles for delivery of sgRNA or shRNA vectors. | psPAX2 (packaging) and pMD2.G (VSV-G envelope). |
| Next-Generation Sequencing (NGS) Kit | For preparation of sequencing libraries from PCR-amplified sgRNA constructs post-screen. | Illumina Nextera XT or custom primer-based amplification kits. |
| Viability/Proliferation Assay Kit | Quantifies cell health or number as a primary readout for essentiality screens or knockdown validation. | CellTiter-Glo (ATP-based, Promega) for 384/96-well plates. |
| CRISPR Clean Control sgRNA | Non-targeting sgRNA that does not match the host genome, used as a negative control for Cas9 activity. | Important for distinguishing generic DNA damage responses from on-target effects. |
The inherent strengths and limitations of RNAi and CRISPR-Cas9 knockout are direct consequences of their first principles. RNAi, acting at the RNA level, offers reversible, titratable knockdown suitable for acute studies and probing essential gene functions but is confounded by incomplete efficiency and off-target transcriptional responses. CRISPR-Cas9, acting at the DNA level, creates permanent, complete knockouts with higher specificity and is the superior tool for generating stable models and definitive loss-of-function data in genome-wide screens, albeit with risks of genomic scarring and DNA damage responses. Within the broader thesis of functional genomics research, the platforms are complementary. A strategic approach often employs CRISPR knockout for definitive target identification and validation, while RNAi is leveraged for follow-on studies of phenotype kinetics or dosage effects. The informed researcher must align the choice of platform with the specific biological question, experimental timeline, and required level of evidence.
From Design to Data: Step-by-Step Methodologies and Application Scenarios
This technical guide provides a comparative overview of CRISPR knockout (CRISPR-KO) and RNA interference (RNAi) workflows for functional genomics, framed within a broader thesis evaluating their respective merits in gene function studies and drug target identification. The choice between these technologies hinges on the specific research question, as CRISPR-KO provides permanent, DNA-level gene ablation, while RNAi offers reversible, transcript-level knockdown, each with distinct implications for phenotypic analysis.
RNAi (RNA Interference): Utilizes short interfering RNA (siRNA) or short hairpin RNA (shRNA) to guide the RNA-induced silencing complex (RISC) to complementary mRNA transcripts, leading to their degradation or translational inhibition. This results in transient or stable knockdown of gene expression.
CRISPR-KO (CRISPR-Cas9 Knockout): Employs a single guide RNA (sgRNA) to direct the Cas9 nuclease to a specific genomic locus, where it induces a double-strand break (DSB). Repair via error-prone non-homologous end joining (NHEJ) leads to small insertions or deletions (indels), often resulting in frameshift mutations and permanent gene disruption.
Key Experimental Workflows
The fundamental workflows for both technologies share common stages but differ critically in execution and biological mechanism.
Detailed Methodologies
Protocol 1: RNAi via Transient siRNA Transfection
- Design: Select 2-3 independent siRNA duplexes targeting different exonic regions of the target mRNA using validated algorithms.
- Reverse Transfection: Seed cells in a 96-well plate (e.g., 3000 cells/well for HeLa). Complex siRNA (final concentration 10-50 nM) with lipid-based transfection reagent in serum-free medium. Incubate 20 min, then add directly to cells.
- Incubation: Assay cells 48-96 hours post-transfection. Include non-targeting siRNA (scramble) and untreated controls.
- Knockdown Validation: Harvest cells for total RNA extraction. Perform reverse transcription and quantitative PCR (qPCR) using TaqMan probes against the target. Normalize to housekeeping genes (GAPDH, ACTB). Calculate % knockdown relative to scramble control.
- Phenotypic Analysis: Proceed with downstream assays (e.g., CellTiter-Glo for viability, caspase-3 assay for apoptosis, or immunostaining).
Protocol 2: CRISPR-KO via Lentiviral Delivery & Clonal Selection
- sgRNA Design & Cloning: Design 2-3 sgRNAs targeting early constitutive exons. Clone oligonucleotides into a lentiviral vector (e.g., lentiCRISPRv2) expressing both sgRNA and Cas9.
- Virus Production: Co-transfect the transfer plasmid with packaging plasmids (psPAX2, pMD2.G) into HEK293T cells using polyethylenimine (PEI). Harvest lentiviral supernatant at 48 and 72 hours.
- Transduction & Selection: Transduce target cells at low MOI (<0.3) with polybrene (8 µg/mL). Select with appropriate antibiotic (e.g., puromycin, 1-2 µg/mL) for 5-7 days.
- Validation of Editing: Extract genomic DNA from the polyclonal population. Amplify target region by PCR. Assess indel frequency via T7 Endonuclease I (T7E1) assay or Sanger sequencing analyzed by Inference of CRISPR Edits (ICE).
- Clonal Isolation: Serially dilute cells to 0.5 cells/well in a 96-well plate. Expand single-cell clones over 2-3 weeks. Screen clones by genomic PCR and Sanger sequencing to identify homozygous frameshift mutants.
- Phenotypic Analysis: Characterize validated knockout clones alongside parental controls.
Signaling Pathway Impact
The mechanistic differences between RNAi and CRISPR-KO lead to distinct biological outcomes, particularly in dynamic signaling networks.
Quantitative Comparison Data
Table 1: Core Technical Parameters
| Parameter | RNAi (siRNA) | CRISPR-KO (Lentiviral) |
|---|---|---|
| Target | mRNA transcript | Genomic DNA |
| Mechanism | Post-transcriptional silencing | NHEJ-mediated frameshift |
| Onset of Effect | 24-48 hours | 24-72 hours (protein depletion after cell division) |
| Duration | Transient (5-7 days) | Permanent, heritable |
| Typical Efficiency | 70-95% knockdown | Varies; often 50-90% indel rate (polyclonal) |
| Key Off-Target Risk | Seed-region mediated miRNA-like effects; saturation of endogenous machinery | sgRNA-dependent cleavage at homologous genomic sites |
| Primary Applications | Acute knockdown, rapid screening, essential gene analysis, in vivo modulation | Complete functional ablation, generation of stable cell lines, genetic modeling |
Table 2: Practical Workflow Considerations
| Consideration | RNAi | CRISPR-KO |
|---|---|---|
| Design Complexity | Moderate (avoid SNPs, secondary structure) | High (requires specific PAM, predict on/off-target) |
| Experimental Timeline | Faster (results in days) | Slower (clonal validation takes weeks) |
| Cost per Gene (Reagents) | Lower | Higher (especially for clonal generation) |
| Phenotypic Penetrance | Incomplete, variable | Complete, uniform in clonal populations |
| Adaptability for Screens | High (arrayed or pooled siRNA libraries) | High (arrayed or pooled sgRNA libraries) |
| Compensation Risk | Higher (protein turnover may allow adaptation) | Lower (complete removal of genetic template) |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Their Functions
| Reagent / Material | Function in RNAi | Function in CRISPR-KO |
|---|---|---|
| Lipofectamine RNAiMAX | Lipid-based transfection reagent optimized for high-efficiency siRNA delivery with low cytotoxicity. | Not typically used for plasmid DNA. |
| FuGENE HD | Useful for plasmid DNA transfection; can be used for shRNA vector delivery. | Often used for transfection of CRISPR plasmids or RNP complexes. |
| Lentiviral Packaging Mix (psPAX2, pMD2.G) | For production of lentiviral particles encoding shRNAs for stable integration. | For production of lentiviral particles encoding Cas9 and sgRNA. |
| Polybrene (Hexadimethrine bromide) | Enhances lentiviral transduction efficiency by neutralizing charge repulsion. | Same function: enhances lentiviral transduction. |
| Puromycin Dihydrochloride | Selective antibiotic for cells transduced with lentiviral vectors carrying puromycin resistance. | Same function: selects for cells expressing Cas9/sgRNA constructs. |
| T7 Endonuclease I (T7E1) | Not applicable. | Detects mismatches in heteroduplex DNA PCR products, indicating indel mutations. |
| Alt-R S.p. Cas9 Nuclease V3 | Not applicable. | High-fidelity, recombinant Cas9 protein for ribonucleoprotein (RNP) complex delivery, reducing off-targets. |
| SYBR Green / TaqMan Assay | Critical for quantifying mRNA knockdown levels via qRT-PCR. | Used to validate knockout by assaying for loss of mRNA (but DNA-level validation is primary). |
| CellTiter-Glo Luminescent Assay | Measures cell viability/proliferation as a primary phenotypic readout post-knockdown. | Measures cell viability/proliferation in knockout clones or populations. |
| RIPA Buffer & Protease Inhibitors | For protein extraction to validate knockdown via western blot. | For protein extraction to confirm absence of target protein in knockout lines. |
Within functional genomics research, the choice between CRISPR-mediated knockout and RNA interference (RNAi) is foundational. CRISPR-Cas9 utilizes a guide RNA (gRNA) to direct DNA cleavage, resulting in permanent gene knockout. RNAi employs small interfering RNA (siRNA) or short hairpin RNA (shRNA) to trigger mRNA degradation, leading to transient gene knockdown. The success of either approach hinges on the precise design of these RNA guides. This technical guide contrasts the design rules, tools, and protocols for gRNA and siRNA/shRNA, framed within the thesis of selecting the optimal tool for robust functional genomics.
Part 1: Core Design Principles and Rules
gRNA Design for CRISPR-Cas9 Knockout
The primary goal is to design a single guide RNA (sgRNA) that directs Cas9 to create a double-strand break (DSB) in the target genomic DNA with high efficiency and minimal off-target effects.
Key Rules:
- Protospacer Adjacent Motif (PAM): The target site must be adjacent to a PAM sequence. For the commonly used Streptococcus pyogenes Cas9 (SpCas9), the PAM is 5'-NGG-3' on the genomic DNA strand complementary to the non-guide strand of the sgRNA.
- Guide Sequence Length: Typically 20 nucleotides upstream of the PAM.
- On-Target Efficiency: Favored sequences have a GC content between 40-60%, avoid long stretches of single nucleotides, and are often located in the early coding exons of the target gene to maximize chances of frameshift-inducing indels.
- Off-Target Minimization: The guide sequence should be unique. Avoid sequences with ≤3 mismatches to other genomic sites, especially in the "seed" region (positions 1-12 closest to the PAM).
- Genomic Context: Target open chromatin regions for better accessibility. Avoid sequences with significant single nucleotide polymorphisms (SNPs).
siRNA/shRNA Design for RNAi Knockdown
The goal is to design a ~21-23 bp duplex (siRNA) or a corresponding expressed hairpin (shRNA) that is incorporated into the RISC complex to cleave complementary mRNA.
Key Rules:
- Target Region: Typically within the coding sequence (CDS) or 3' UTR of the mRNA. Avoid regions near the start codon or within complex secondary structures.
- Sequence Characteristics: Optimal GC content of ~30-50%. Avoid extreme GC content.
- Specificity: Use BLAST to ensure minimal homology with other transcripts. The "seed" region (positions 2-8 from the 5' end of the guide strand) is critical for specificity.
- Asymmetry Rule (RISC Loading): Design the duplex so the intended guide strand has a thermodynamically less stable 5' end relative to the passenger strand, promoting its loading into RISC.
- Chemical Modifications: For therapeutic siRNA, introduce chemical modifications (e.g., 2'-O-methyl, phosphorothioate) to enhance stability and reduce immunogenicity.
Part 2: Quantitative Data Comparison
Table 1: Core Design Parameter Comparison
| Parameter | CRISPR gRNA | siRNA/shRNA |
|---|---|---|
| Target Molecule | Genomic DNA | Messenger RNA (mRNA) |
| Mechanism | DNA cleavage (DSB) | mRNA cleavage or translational repression |
| Typical Length | 20 nt spacer + scaffold | 21-23 bp duplex |
| Key Motif | PAM (e.g., NGG for SpCas9) | Seed region (positions 2-8) |
| Primary Goal | Maximize on-target cleavage, minimize DNA off-targets | Maximize mRNA degradation, minimize seed-based off-targets |
| Effect Duration | Permanent (knockout) | Transient (knockdown) |
| Key Design Tools | CRISPick, ChopChop, Benchling | Dharmacon siRNA Design, siDirect, BLOCK-iT |
Table 2: Performance Metrics in Functional Genomics
| Metric | CRISPR Knockout | RNAi Knockdown |
|---|---|---|
| Gene Suppression Level | Typically >90% (complete loss of function) | Variable, often 70-90% (partial reduction) |
| Phenotype Penetrance | High (biallelic disruption) | Can be dose-dependent and incomplete |
| Off-Target Effect Type | DNA-level indels at homologous sites | RNA-level; seed-driven miRNA-like repression |
| Time to Effect | Slower (requires DNA repair and protein turnover) | Rapid (hours to days post-transfection) |
| Common Validation | Sanger/NGS for indels, Western blot for protein loss | qRT-PCR for mRNA, Western blot for protein reduction |
Part 3: Experimental Protocols
Protocol 1: Validating gRNA On-Target Efficiency (T7 Endonuclease I Assay)
This protocol assesses the rate of indel formation at the target locus.
Detailed Methodology:
- Cell Transfection: Transfect cells (e.g., HEK293T) with your Cas9/gRNA plasmid or RNP complex using an appropriate method (lipofection, electroporation).
- Genomic DNA Extraction: 72 hours post-transfection, harvest cells and extract genomic DNA using a commercial kit.
- PCR Amplification: Design primers ~200-400 bp flanking the target site. Perform PCR to amplify the target region from the extracted gDNA.
- DNA Denaturation & Reannealing: Purify the PCR product. Mix 200 ng of the product in 1X NEBuffer 2, denature at 95°C for 5 min, then slowly reanneal by ramping down to 25°C at -0.1°C/sec. This forms heteroduplex DNA if indels are present.
- T7EI Digestion: Add 0.5 µL of T7 Endonuclease I enzyme to the reannealed DNA and incubate at 37°C for 15-30 minutes. T7EI cleaves mismatched heteroduplexes.
- Analysis: Run the digested product on a 2% agarose gel. Compare to an undigested control. Cleavage into two smaller bands indicates successful genome editing. The indel frequency can be estimated from band intensities.
Protocol 2: Validating siRNA/shRNA Knockdown Efficiency (qRT-PCR and Western Blot)
This two-part protocol measures reduction in target mRNA and protein levels.
Detailed Methodology: A. mRNA Level (qRT-PCR):
- Cell Transfection: Transfect cells with siRNA or shRNA-expressing vector.
- RNA Isolation: 24-48 hours post-transfection, lyse cells and isolate total RNA using a kit (e.g., TRIzol or column-based). Treat with DNase I.
- cDNA Synthesis: Perform reverse transcription using 0.5-1 µg of total RNA, oligo(dT) or random hexamer primers, and a reverse transcriptase enzyme.
- Quantitative PCR: Prepare reactions with cDNA template, gene-specific primers, and SYBR Green master mix. Run in a real-time PCR cycler. Include a housekeeping gene (e.g., GAPDH, ACTB) for normalization.
- Data Analysis: Calculate ∆∆Ct values relative to a non-targeting control siRNA-treated sample to determine the fold reduction in mRNA.
B. Protein Level (Western Blot):
- Protein Extraction: 48-72 hours post-transfection, lyse cells in RIPA buffer containing protease inhibitors. Centrifuge to clear debris and quantify protein concentration.
- Gel Electrophoresis: Load 20-30 µg of protein per lane on an SDS-PAGE gel. Run and then transfer proteins to a PVDF membrane.
- Immunodetection: Block the membrane, then incubate with primary antibody against the target protein, followed by an HRP-conjugated secondary antibody.
- Visualization: Use chemiluminescent substrate and image the blot. Re-probe for a loading control (e.g., β-Actin, GAPDH). Densitometry analysis quantifies protein knockdown.
Part 4: Visualizations
Title: gRNA Design and Validation Workflow
Title: RNAi vs. CRISPR Molecular Pathways
Part 5: The Scientist's Toolkit
Table 3: Research Reagent Solutions for Guide Design & Validation
| Reagent / Material | Function | Application |
|---|---|---|
| T7 Endonuclease I | Enzyme that cleaves mismatched heteroduplex DNA. | Detecting indel mutations in CRISPR-edited pools (Protocol 1). |
| SYBR Green qPCR Master Mix | Fluorescent dye that binds double-stranded DNA for real-time quantification. | Measuring mRNA levels in siRNA knockdown experiments (Protocol 2). |
| RIPA Lysis Buffer | A buffer for efficient cell lysis and total protein extraction. | Preparing samples for Western blot analysis post-knockdown/knockout. |
| Lipofectamine 3000 | A lipid-based transfection reagent for nucleic acids. | Delivering plasmid DNA (Cas9/sgRNA) or siRNA into mammalian cells. |
| Dharmacon ON-TARGETplus siRNA | Pre-designed, chemically modified siRNAs with verified reduced off-target effects. | High-confidence RNAi knockdown studies. |
| Alt-R S.p. Cas9 Nuclease V3 | High-purity, recombinant SpCas9 protein for RNP complex formation. | CRISPR editing with rapid kinetics and reduced off-targets compared to plasmid delivery. |
| Next-Generation Sequencing (NGS) Kit | For high-throughput sequencing of amplicons. | Comprehensive off-target profiling and precise quantification of editing efficiency. |
Within functional genomics research, the choice between CRISPR-Cas9-mediated knockout and RNA interference (RNAi) for gene silencing is pivotal. The efficacy, specificity, and outcome of these techniques are fundamentally governed by the delivery system used to introduce genetic material into target cells. This technical guide provides an in-depth analysis of three core delivery platforms—Lentivirus, Lipid Nanoparticles (LNPs), and Electroporation—as applied to both CRISPR and RNAi workflows. The selection of an appropriate delivery method directly influences key thesis parameters such as knockout efficiency versus knockdown kinetics, off-target effects, and applicability across diverse cell types.
Core Delivery Technologies: Mechanisms and Applications
Lentiviral Delivery
Lentivirus, a subclass of retroviruses, enables stable genomic integration of delivered constructs. For RNAi, this typically involves short hairpin RNA (shRNA) expression cassettes. For CRISPR-Cas9, lentivectors can deliver single-guide RNA (sgRNA) alone (in Cas9-expressing cells) or both Cas9 and sgRNA as separate or combined constructs.
Key Mechanism: The lentiviral capsid facilitates cell entry via receptor-mediated endocytosis. Upon uncoating, the viral RNA is reverse-transcribed into DNA, which integrates into the host genome, leading to long-term, stable expression of the RNAi or CRISPR components.
Advantages: High transduction efficiency in difficult cells (e.g., primary, non-dividing), stable long-term expression. Disadvantages: Limited cargo capacity (~8-10 kb), risk of insertional mutagenesis, variable integration copy number, biosafety level 2 requirements.
Lipid Nanoparticle (LNP) Delivery
LNPs are synthetic, non-viral carriers that encapsulate nucleic acids (siRNA for RNAi; sgRNA and/or Cas9 mRNA for CRISPR). They are the leading platform for in vivo therapeutic delivery.
Key Mechanism: Cationic or ionizable lipids form a protective shell around the nucleic acid payload. The LNP fuses with the cell membrane or is taken up via endocytosis. In the endosome, ionizable lipids become positively charged at low pH, disrupting the endosomal membrane and releasing the payload into the cytoplasm.
Advantages: High delivery efficiency in vivo, low immunogenicity compared to viruses, no genomic integration, scalable manufacturing. Disadvantages: Transient expression, potential cytotoxicity at high doses, complexity in formulation.
Electroporation
Electroporation uses controlled electrical pulses to create transient pores in the cell membrane, allowing nucleic acids or ribonucleoproteins (RNPs) to enter the cytoplasm directly.
Key Mechanism: Application of an external electric field exceeds the capacitance of the cell membrane, inducing the formation of hydrophilic pores. Molecules in the surrounding buffer diffuse into the cell. Pores reseal once the pulse ceases.
Advantages: Applicable to a wide range of molecules (DNA, RNA, RNP), high efficiency in hard-to-transfect cells (e.g., immune cells, stem cells), no cargo size limit for CRISPR RNP delivery. Disadvantages: Can cause significant cell stress and mortality, requires specialized equipment, less suited for in vivo systemic delivery.
Quantitative Comparison of Delivery Systems
Table 1: Comparative Analysis of Delivery Systems for CRISPR and RNAi
| Parameter | Lentivirus | Lipid Nanoparticles (LNPs) | Electroporation |
|---|---|---|---|
| Primary Cargo (RNAi) | shRNA-encoding plasmid | siRNA | siRNA |
| Primary Cargo (CRISPR) | sgRNA ± Cas9 DNA | Cas9 mRNA + sgRNA or RNP | Cas9-sgRNA RNP or plasmids |
| Delivery Efficiency* | High (70-95%) | Moderate to High (50-90%) | Very High (80-95%) |
| Expression Kinetics | Slow, stable (days to weeks) | Fast, transient (peak at 24-48h) | Immediate, transient (RNP) or delayed (DNA) |
| Genomic Integration | Yes (random) | No | No (RNP); Yes (if DNA plasmid) |
| Typical Use Case | Stable cell line generation, in vivo long-term studies | In vivo systemic delivery, difficult cells in vitro | Primary cells, immune cells, hard-to-transfect lines |
| Throughput | Moderate | High | Low to Moderate (well-based systems enable higher) |
| Cytotoxicity Risk | Low (but mutagenesis risk) | Moderate | High |
| Cost & Complexity | High (production, biosafety) | Moderate to High (formulation) | Low (reagents), High (equipment) |
Efficiency is cell-type dependent; ranges are representative for common mammalian cell lines.
Table 2: Suitability for Functional Genomics Thesis Parameters
| Thesis Consideration | Lentivirus | LNPs | Electroporation |
|---|---|---|---|
| CRISPR Knockout Permanence | Excellent (stable integration) | Poor (transient) | Good (RNP: transient but efficient) |
| RNAi Knockdown Duration | Excellent (stable shRNA expression) | Good (siRNA: 5-7 days) | Good (siRNA: 5-7 days) |
| On/Off-Target Specificity | Lower (random integration effects) | Higher (no integration, transient) | Highest (RNP: rapid degradation minimizes off-target) |
| Screening Scalability | Excellent for pooled screens | Good for arrayed screens | Moderate for arrayed screens |
Detailed Experimental Protocols
Protocol 4.1: Lentiviral Production and Transduction for shRNA or CRISPR
Objective: Produce VSV-G pseudotyped lentivirus and transduce target cells. Materials: Packaging plasmids (psPAX2, pMD2.G), transfer plasmid (shRNA or CRISPR), HEK293T cells, PEI transfection reagent, growth media, polybrene (8 µg/mL). Procedure:
- Day 1: Seed HEK293T cells in a 10 cm dish to reach 70-80% confluency the next day.
- Day 2: Co-transfect using PEI: Mix 10 µg transfer plasmid, 7.5 µg psPAX2, and 2.5 µg pMD2.G in 500 µL serum-free media. Add 60 µL PEI (1 mg/mL), vortex, incubate 15 min at RT. Add dropwise to cells.
- Day 3: Replace medium with fresh complete medium.
- Day 4 & 5: Harvest viral supernatant at 48h and 72h post-transfection. Filter through a 0.45 µm PES filter. Aliquot and store at -80°C or concentrate via ultracentrifugation.
- Transduction: Plate target cells. Add viral supernatant and polybrene. Centrifuge at 800-1000 x g for 30-60 min (spinoculation). Replace media after 24h. Apply selection (e.g., puromycin) 48h post-transduction.
Protocol 4.2: LNP Formulation and Transfection for siRNA or CRISPR RNP
Objective: Formulate LNPs encapsulating siRNA or Cas9 RNP via microfluidic mixing. Materials: Ionizable lipid (e.g., DLin-MC3-DMA), cholesterol, DSPC, DMG-PEG-2000, nucleic acid payload, 25 mM sodium acetate buffer (pH 4.0), PBS (pH 7.4), microfluidic mixer. Procedure:
- Prepare Lipid Mix: Dissolve ionizable lipid, cholesterol, DSPC, and PEG-lipid at a molar ratio (e.g., 50:38.5:10:1.5) in ethanol.
- Prepare Aqueous Phase: Dilute siRNA or Cas9 RNP in sodium acetate buffer (pH 4.0).
- Mixing: Using a staggered herringbone microfluidic device, rapidly mix the lipid phase and aqueous phase at a 3:1 flow rate ratio (aqueous:lipid). Total flow rate ~12 mL/min.
- Dialysis/Buffer Exchange: Dialyze the formed LNP suspension against PBS (pH 7.4) for 2 hours using a Slide-A-Lyzer cassette (MWCO 20KDa) to remove ethanol and adjust pH.
- Characterization: Measure particle size (target ~80-100 nm) via DLS and encapsulation efficiency (>90%) using a Ribogreen assay.
- Transfection: Add LNPs directly to cells in culture at an appropriate dose (e.g., 0.5-1 µM siRNA final concentration). Assay after 24-72h.
Protocol 4.3: Electroporation of Cas9 RNP or siRNA
Objective: Deliver Cas9-sgRNA RNP or siRNA into suspension cells (e.g., Jurkat, primary T cells). Materials: Neon Electroporation System or comparable, electroporation buffer, Cas9 protein, synthetic sgRNA, siRNA, sterile tips, 24-well plate. Procedure:
- RNP Complex Formation: For CRISPR, incubate 10 µg (≈60 pmol) recombinant Cas9 protein with 5 µg (≈120 pmol) sgRNA in a total volume of 10 µL for 10-20 min at room temperature.
- Cell Preparation: Harvest and wash 5 x 10^5 to 1 x 10^6 cells twice in PBS. Resuspend cells in the recommended electroporation buffer (e.g., Buffer R for Neon) to a concentration of 1-5 x 10^7 cells/mL.
- Electroporation Setup: Mix 10 µL of cell suspension with 10 µL of RNP complex or 5 µL of siRNA (5 µM stock). Aspirate into a Neon tip.
- Pulse Conditions: Apply optimized electrical pulse (e.g., for Jurkat: 1320V, 10ms, 3 pulses). Immediately transfer electroporated cells to pre-warmed complete medium in a 24-well plate.
- Post-Processing: Culture cells and assay for knockout (via T7E1 or NGS) or knockdown (via qPCR) after 48-96 hours.
Visualization of Workflows and Mechanisms
Title: Intracellular LNP Pathway for RNAi and CRISPR
Title: Decision Tree for Functional Genomics Delivery
Title: Integrated CRISPR and RNAi Delivery Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents for Delivery Experiments
| Reagent / Material | Function / Application | Example Vendor/Product |
|---|---|---|
| Lentiviral Packaging Plasmids | Provide viral structural (gag/pol) and envelope (VSV-G) proteins in trans for safe replication-incompetent virus production. | Addgene: psPAX2, pMD2.G |
| Polyethylenimine (PEI), Linear | Cationic polymer for transient transfection of packaging plasmids into producer cells (e.g., HEK293T). | Polysciences, JetPEI |
| Ionizable Cationic Lipid | Key component of LNPs; promotes encapsulation and endosomal escape. Critical for in vivo efficacy. | MedChemExpress (DLin-MC3-DMA), Avanti Lipids |
| DMG-PEG-2000 | PEGylated lipid used in LNP formulation to reduce aggregation, improve stability, and prolong circulation half-life. | Avanti Polar Lipids |
| Recombinant Cas9 Nuclease | High-purity protein for RNP formation and electroporation, minimizing DNA-based delivery artifacts. | Thermo Fisher, IDT, Sino Biological |
| Chemically Modified Synthetic sgRNA/siRNA | Enhanced stability and reduced immunogenicity; essential for LNP and electroporation workflows. | Dharmacon, IDT, Sigma-Aldrich |
| Neon Transfection System / Nucleofector | Specialized electroporation devices with optimized buffers and protocols for high-efficiency delivery to sensitive cells. | Thermo Fisher, Lonza |
| Polybrene (Hexadimethrine bromide) | Cationic polymer used to enhance lentiviral transduction efficiency by neutralizing charge repulsion. | Sigma-Aldrich |
| Puromycin Dihydrochloride | Selection antibiotic for cells transduced with lentiviral vectors containing a puromycin resistance gene. | Thermo Fisher, InvivoGen |
| Ribogreen / PicoGreen Assay Kits | Fluorescent nucleic acid stains for quantifying encapsulation efficiency of LNPs. | Thermo Fisher |
The selection between CRISPR-Cas9-mediated knockout and RNA interference (RNAi)-mediated knockdown is a foundational decision in functional genomics and drug target validation. While both aim to reduce gene function, their mechanisms, temporal dynamics, and outcomes differ substantially, making each uniquely suited for specific applications.
CRISPR-Cas9 creates permanent, DNA-level double-strand breaks, leading to frameshift mutations and complete gene knockout. Its high specificity and permanence make it ideal for large-scale genetic screens to identify essential genes and long-term phenotypic studies. However, it is slower to implement due to the need for DNA cleavage and turnover.
RNAi utilizes small interfering RNA (siRNA) or short hairpin RNA (shRNA) to degrade target mRNA or inhibit its translation, resulting in transient knockdown. Its rapid effect (hours to days) is optimal for acute functional assays and rapid target validation, though off-target effects and incomplete silencing are notable limitations.
The core thesis guiding this whitepaper is: CRISPR knockout is the superior tool for definitive, genome-wide loss-of-function screens, while RNAi is optimal for rapid, flexible knockdown in acute-phase target validation and signaling pathway deconvolution.
Quantitative Comparison: CRISPR vs. RNAi
Table 1: Core Characteristics of CRISPR-Cas9 and RNAi
| Parameter | CRISPR-Cas9 (Knockout) | RNAi (Knockdown) |
|---|---|---|
| Molecular Target | Genomic DNA | mRNA |
| Mechanism | DSB, NHEJ/HDR | RISC-mediated mRNA cleavage/translational inhibition |
| Effect Type | Permanent knockout | Transient knockdown |
| Onset of Effect | Days (requires protein turnover) | Hours to days |
| Duration of Effect | Stable, heritable | Transient (typically 3-7 days) |
| Primary Risk | Off-target DNA cleavage, genomic rearrangements | Off-target transcriptional effects, seed-based mimicry |
| Typical Efficiency | High (often >80% indel rate) | Variable (40-90% mRNA reduction) |
| Ideal Application | Genome-wide/pooled screens, essential gene identification, long-term phenotype studies | Rapid target validation, signaling pathway analysis, acute phenotype assessment |
Table 2: Performance Metrics in Key Applications (Recent Benchmarking Data)
| Application | Best Tool | Key Metric (Typical Result) | Rationale for Superiority |
|---|---|---|---|
| Genome-wide Loss-of-function Screen | CRISPR | Hit reproducibility (Pearson r > 0.8 between replicates) | Higher specificity, lower false-negative rate for essential genes. |
| Rapid Kinase Target Validation | RNAi | Phenotype onset (< 72 hrs post-transfection) | Faster protein depletion without waiting for genomic editing. |
| Synthetic Lethality Screening | CRISPR | Z'-factor for screen quality (>0.5) | Clearer, more complete elimination of gene function. |
| Signaling Pathway Deconvolution | RNAi | Dose-responsive phenotype correlation (R² > 0.7) | Tunable knockdown levels allow titration of signal. |
Detailed Methodologies
Protocol: CRISPR-Cas9 Pooled Library Screen for Essential Genes
This protocol identifies genes essential for cell proliferation/survival.
Materials:
- Cells: Preferably near-diploid, rapidly dividing cell line (e.g., K562, A549).
- CRISPR Library: Brunello or similar genome-wide sgRNA library (4-6 sgRNAs/gene, ~80,000 total sgRNAs).
- Lentiviral Packaging System: psPAX2, pMD2.G, and transfection reagent (e.g., PEI).
- Culture Media: Appropriate complete medium, plus puromycin for selection.
- PCR & NGS Reagents: Primers for amplifying integrated sgRNA sequences, High-Fidelity polymerase, NGS cleanup kits.
Procedure:
- Library Amplification & Lentivirus Production: Transform the sgRNA plasmid library into competent E. coli and amplify to maintain >200x coverage. Co-transfect HEK293T cells with the library plasmid, psPAX2, and pMD2.G to produce lentivirus. Harvest supernatant at 48 and 72 hours.
- Cell Infection & Selection: Infect target cells at a low MOI (~0.3) to ensure most cells receive one sgRNA. Include a non-targeting control sgRNA. 24-48 hours post-infection, select with puromycin (e.g., 1-2 µg/mL) for 3-5 days.
- Screen Passage & Harvest: Passage cells, maintaining a representation of >500 cells per sgRNA at all times. Harvest genomic DNA from a minimum of 50 million cells at the initial time point (T0) and at the end point (T-end, typically 14-21 population doublings).
- sgRNA Amplification & Sequencing: PCR-amplify integrated sgRNA cassettes from gDNA using barcoded primers. Pool PCR products and perform NGS (MiSeq/NextSeq) to a depth of >200 reads per sgRNA.
- Data Analysis: Align reads to the sgRNA library reference. Using a tool like MAGeCK, calculate sgRNA depletion/enrichment by comparing read counts at T-end vs. T0. Rank genes by robust scoring algorithms (e.g., MAGeCK RRA).
Protocol: RNAi-Mediated Rapid Knockdown for Drug Target Validation
This protocol validates a putative drug target by phenocopying a drug's effect with targeted gene knockdown.
Materials:
- Cells: Relevant disease model cell line.
- siRNA: Validated ON-TARGETplus siRNA pools (Dharmacon) or individual siRNAs targeting the gene of interest. Include non-targeting (scramble) and positive control (e.g., PLK1) siRNAs.
- Transfection Reagent: Lipid-based (e.g., Lipofectamine RNAiMAX) or electroporation system.
- Assay Reagents: Cell viability/cytotoxicity assay (e.g., CellTiter-Glo), Western blot or qRT-PCR materials for knockdown confirmation.
Procedure:
- Reverse Transfection: Seed cells in 96-well plates. Complex siRNA (final concentration 10-25 nM) with RNAiMAX in Opti-MEM and add directly to cells. Incubate for 48-96 hours.
- Knockdown Confirmation: At 48-72 hours, harvest parallel wells for mRNA (qRT-PCR) or protein (Western blot) to confirm target reduction (>70% knockdown is ideal).
- Phenotypic Assessment: At the desired time point (e.g., 72h for proliferation, 96h for a complex phenotype), perform the relevant assay (e.g., add CellTiter-Glo, measure luminescence).
- Data Integration & Validation: Compare phenotype in target-knockdown wells to both non-targeting and positive controls. A successful validation occurs when target knockdown phenocopies the effect of the drug targeting the same protein (e.g., similar reduction in cell viability). Use combination studies (sub-optimal drug dose + sub-optimal knockdown) to assess synergy.
Visualizations
(Diagram 1: RNAi Mechanism & Workflow for Rapid Knockdown)
(Diagram 2: CRISPR Pooled Screening Workflow for Gene Discovery)
(Diagram 3: Decision Logic for Selecting CRISPR vs. RNAi)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR and RNAi Workflows
| Category | Reagent/Kit | Primary Function | Key Consideration |
|---|---|---|---|
| CRISPR Screening | Brunello/GeCKO v2 sgRNA Library | Genome-wide, high-coverage sgRNA sets for knockout screens. | Optimized for reduced off-target effects. Maintain high representation during amplification. |
| CRISPR Delivery | Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Produces VSV-G pseudotyped lentivirus for stable sgRNA integration. | Use 2nd/3rd generation systems for enhanced safety and titer. |
| RNAi Reagents | ON-TARGETplus siRNA (Dharmacon) | Chemically modified siRNA pools for specific, potent knockdown with reduced off-targets. | Use SMARTpool designs (4 siRNAs/gene) for reliable results. |
| Transfection | Lipofectamine RNAiMAX (Invitrogen) | Lipid-based reagent optimized for high-efficiency siRNA delivery with low cytotoxicity. | Perform reverse transfections for best consistency in adherent cells. |
| Knockdown Validation | TaqMan Gene Expression Assays (Applied Biosystems) | Probe-based qRT-PCR for absolute quantification of target mRNA remaining post-knockdown. | More specific than SYBR Green; requires primer-probe set design. |
| Phenotypic Readout | CellTiter-Glo Luminescent Assay (Promega) | Quantifies ATP as a biomarker for metabolically active cells in viability/proliferation assays. | Homogeneous "add-mix-measure" format, ideal for high-throughput. |
| NGS Library Prep | NEBNext Ultra II DNA Library Prep Kit | Prepares sequencing libraries from PCR-amplified sgRNA sequences. | Includes bead-based size selection for consistent insert size. |
| Screen Analysis | MAGeCK (Bioinformatics Tool) | Statistical model to rank essential genes from CRISPR screen NGS count data. | Accounts for multiple sgRNAs per gene and screen variance. |
This technical guide evaluates the application of CRISPR-Cas9 knockout (KO) and RNA interference (RNAi) screening in functional genomics across three critical disease areas. The core thesis posits that while RNAi remains a valuable tool for transient knockdown and studying essential genes, CRISPR-KO offers superior specificity, penetrance, and durability for loss-of-function studies, driving its dominance in definitive target identification and validation workflows. The following case studies, supported by current data and protocols, illustrate this paradigm.
Case Study 1: Oncology – Identifying Synthetic Lethal Interactions in BRCA-Deficient Cancers
Thesis Context: CRISPR-KO screens are indispensable for uncovering synthetic lethal partners due to complete gene ablation, whereas RNAi's incomplete knockdown can obscure these phenotypes.
Experimental Protocol:
- Cell Line Engineering: Generate isogenic pairs of BRCA1-proficient and -deficient human ovarian cancer cell lines (e.g., using CRISPR-KO of BRCA1).
- Library Transduction: Transduce each cell line with a genome-wide CRISPR-KO lentiviral sgRNA library (e.g., Brunello) at a low MOI to ensure single integration. Perform parallel screens with a genome-wide shRNA library (e.g., TRC) for comparison.
- Selection & Passaging: Select transduced cells with puromycin (shRNA) or puromycin+blasticidin (CRISPR). Passage cells for 14-21 population doublings.
- Sample Collection & Sequencing: Harvest genomic DNA at Day 0 and endpoint. PCR-amplify integrated sgRNA or shRNA barcodes and perform deep sequencing.
- Data Analysis: Use MAGeCK or similar algorithms to calculate differentially enriched or depleted guides between Day 0 and endpoint, and between BRCA1-proficient vs. -deficient conditions.
Key Data Summary:
Table 1: Comparison of CRISPR-KO vs. RNAi Screen for PARP1 Synthetic Lethality
| Metric | CRISPR-KO Screen | Genome-wide shRNA Screen |
|---|---|---|
| Top Hit | PARP1 (sgRNAs highly depleted in BRCA1-KO only) | PARP1 (shRNAs depleted, but signal noisier) |
| Phenotype Penetrance | >99% gene disruption; clear synthetic lethality | ~70-90% knockdown; attenuated lethality phenotype |
| False Positive Rate | Low (minimal off-target effects with optimized sgRNAs) | Moderate (seed-sequence based off-targets common) |
| Validation Rate | Typically >80% for top candidates | Typically 40-60% for top candidates |
| Key Advantage | Definitive identification of non-essential genes in a specific genetic context. | Can knockdown essential genes to study dose-dependent effects. |
Diagram Title: Synthetic Lethality Mechanism of PARP1 in BRCA-Deficient Cells
Case Study 2: Neuroscience – Unraveling Neurodegenerative Disease Pathways in iPSC-Derived Neurons
Thesis Context: CRISPR-KO in isogenic induced pluripotent stem cell (iPSC) lines provides a genetically clean background to study neurodegenerative mechanisms, overcoming RNAi's variable knockdown in differentiated neurons.
Experimental Protocol:
- iPSC Line Generation: Use CRISPR-KO to introduce loss-of-function mutations in the C9orf72 gene in control iPSCs, or correct the hexanucleotide repeat expansion in patient-derived iPSCs, creating isogenic pairs.
- Differentiation: Differentiate iPSCs into cortical glutamatergic neurons using a standardized dual-SMAD inhibition protocol.
- Perturbation & Phenotyping: Perform a focused CRISPR-KO screen targeting genes in the autophagy/lysosomal pathway. Assess neuronal survival (high-content imaging), TDP-43 mislocalization (immunofluorescence), and dipeptide repeat protein levels (Western blot) at 40 and 60 days post-differentiation.
- Functional Rescue: Overexpress top-hit genes (e.g., TMEM106B) from the screen in C9orf72-mutant neurons to confirm pathway involvement.
Key Data Summary:
Table 2: Functional Genomics in iPSC-Derived Neuronal Models
| Aspect | CRISPR-KO in Isogenic iPSCs | RNAi in Differentiated Neurons |
|---|---|---|
| Genetic Background | Perfectly matched isogenic controls. | Variable genetic background between control and patient lines. |
| Perturbation Duration | Stable, permanent KO suitable for long-term neuronal assays. | Transient (days-weeks); knockdown efficacy wanes over time. |
| Phenotype Specificity | High; direct link between genotype and phenotype. | Confounded by incomplete knockdown and genetic variation. |
| Screen Suitability | Excellent for long-term survival and proteopathy screens. | Best for acute signaling or short-term toxicity screens. |
| Key Advantage | Enables causal studies in a human neuronal context over extended timelines. | Faster turnaround for initial hypothesis testing. |
Diagram Title: CRISPR-iPSC Workflow for Neurodegenerative Disease Screening
Case Study 3: Infectious Disease – Host Factor Discovery for Viral Pathogenesis
Thesis Context: CRISPR-KO enables the comprehensive identification of essential host factors for viral entry and replication, with fewer false negatives than RNAi, critical for antiviral target discovery.
Experimental Protocol:
- Cell Line Preparation: Culture permissive cell line (e.g., A549 for influenza, Huh-7 for HCV).
- Genome-Wide Screening: Transduce cells with a human genome-wide CRISPR-KO library. Select and expand.
- Viral Challenge: Infect the pooled KO cell population with a reporter virus (e.g., GFP-expressing influenza A virus) at a low MOI. Include an uninfected control.
- FACS Sorting: At 48-72 hours post-infection, sort cells into GFP-negative (resistant) and GFP-positive (susceptible) populations.
- Sequencing & Analysis: Extract genomic DNA from sorted populations, sequence sgRNA barcodes, and identify sgRNAs enriched in the resistant population (essential host factors).
Key Data Summary:
Table 3: Host Factor Screen for Influenza A Virus (IAV)
| Factor Category | Exemplar Gene (CRISPR-KO Hit) | RNAi Performance | Role in IAV Lifecycle |
|---|---|---|---|
| Entry/Endocytosis | CCDC50 (YOD1) | Identified, but weaker phenotype | Regulates viral uncoating via ubiquitination. |
| Transcripton/Replication | NPTX1 | Missed in multiple screens | Binds viral polymerase, essential for function. |
| Nuclear Trafficking | KPNA4 | Consistently identified | Imports viral ribonucleoprotein into nucleus. |
| Assembly/Egress | VPS29 (Retromer) | Partially identified | Critical for trafficking viral components. |
| Key Advantage of KO | Identifies genes where partial KD is insufficient to block infection (e.g., NPTX1). | Useful for identifying rate-limiting, dosage-sensitive factors. |
Diagram Title: Host Factor Essentiality: Complete KO vs. Partial KD
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Functional Genomics Screens
| Reagent / Material | Function & Description | Example Product/Catalog |
|---|---|---|
| Genome-wide CRISPR-KO Library | Pooled sgRNAs targeting all human genes for loss-of-function screens. | Broad Institute Brunello Library (~74k sgRNAs). |
| Genome-wide shRNA Library | Pooled shRNAs for RNAi-based knockdown screens. | TRC shRNA library (Sigma). |
| Lentiviral Packaging Mix | Plasmid mix (psPAX2, pMD2.G) for producing lentiviral particles. | Addgene #12260 & #12259. |
| Polybrene (Hexadimethrine Bromide) | Enhances lentiviral transduction efficiency. | Sigma H9268. |
| Puromycin Dihydrochloride | Selects for cells successfully transduced with shRNA or CRISPR vectors. | Thermo Fisher A1113803. |
| Blasticidin S HCl | Additional selection agent for CRISPR vectors carrying bsd resistance. | Thermo Fisher A1113903. |
| Next-Gen Sequencing Kit | For preparing sgRNA/shRNA amplicon libraries from genomic DNA. | Illumina Nextera XT. |
| MAGeCK Computational Tool | Statistical model to identify significantly enriched/depleted guides from NGS data. | Open-source software. |
| High-Content Imaging System | Automated microscopy for analyzing complex cellular phenotypes in validation. | PerkinElmer Operetta, ImageXpress Micro. |
Solving Common Pitfalls: Troubleshooting and Optimization Strategies for Reliable Results
Within the ongoing debate regarding CRISPR knockout versus RNAi for functional genomics research, the challenge of off-target effects remains a critical point of comparison. While RNAi is plagued by seed-based off-targeting and incomplete knockdown, CRISPR-Cas9 offers precise DNA cleavage but is susceptible to guide RNA mismatches. A rigorous framework for predicting and validating these unintended effects is therefore essential for confident experimental interpretation and therapeutic development. This guide details the integrated computational and experimental methodologies required to address this core issue.
Computational Prediction of Off-Target Effects
Computational tools predict potential off-target sites by analyzing sequence similarity to the intended target. The algorithms weigh factors like the number, position, and type of mismatches, as well as genomic accessibility.
Key Algorithms and Tools
The following table summarizes leading prediction tools, their underlying algorithms, and primary outputs.
Table 1: Computational Off-Target Prediction Tools
| Tool Name | Core Algorithm/Model | Primary Output | Suited For |
|---|---|---|---|
| CRISPOR | MIT & CFD scoring | Ranked list of off-target sites, primers | sgRNA design & validation |
| Cas-OFFinder | Hamming distance search | Genome-wide potential sites | Any PAM, bulk analysis |
| CCTop | Bowtie2 alignment + scoring | On/off-target predictions with scores | Standard Cas9 & nickases |
| CHOPCHOP | Multiple alignment backends | Visualized targets & off-targets | Design & screening |
| GuideScan2 | Incorporates chromatin data | Off-targets with activity scores | CRISPRa/i & epigenetic context |
Quantitative Comparison of Prediction Metrics
Different scoring systems (e.g., MIT, CFD, Doench '16) prioritize different mismatch characteristics. The table below compares their performance based on recent benchmarking studies.
Table 2: Performance Metrics of Off-Target Scoring Systems
| Scoring System | AUC-ROC (Mean) | Key Weighted Factors | Best Use Case |
|---|---|---|---|
| MIT Score | 0.78 | Positions 1-12, mismatch type | Initial sgRNA filtering |
| CFD Score | 0.85 | Mismatch position & identity | High-specificity design |
| DeepCRISPR | 0.89 | Sequence & epigenetic features | Complex cellular models |
| Elevation | 0.87 | Ensemble of models | Therapeutic development |
Experimental Validation Methodologies
Computational predictions require empirical confirmation. The following protocols detail the most robust validation methods.
Protocol: In Vitro Cleavage Assays (GUIDE-Seq)
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) is a sensitive, unbiased method for detecting double-strand breaks (DSBs) in living cells.
Materials & Workflow:
- Transfection: Co-transfect cells with Cas9/sgRNA RNP and a double-stranded oligodeoxynucleotide (dsODN) tag.
- Integration: The dsODN tag is captured into DSB sites during repair.
- Genomic DNA Extraction & Shearing: Harvest genomic DNA and fragment it.
- Library Prep & Enrichment: Perform adapter ligation and PCR enrichment using primers specific to the integrated dsODN tag.
- Sequencing & Analysis: High-throughput sequencing followed by alignment to the reference genome to identify off-target integration sites.
GUIDE-Seq Experimental Workflow
Protocol: Targeted Amplicon Sequencing for Validation
This targeted method quantifies mutations at predicted off-target loci.
Detailed Steps:
- Design Primers: Design PCR primers (amplicons 200-300 bp) flanking each predicted off-target site and the on-target site.
- PCR Amplification: Amplify loci from treated and control cell genomic DNA using high-fidelity polymerase.
- Attach Barcodes & Adapters: Perform a second, limited-cycle PCR to attach unique dual indices and sequencing adapters.
- Pool & Sequence: Pool barcoded amplicons and sequence on a high-coverage platform (e.g., Illumina MiSeq).
- Analysis: Use pipelines (CRISPResso2, Cas-Analyzer) to align reads and quantify insertion/deletion (indel) frequencies at each locus.
Targeted Amplicon Sequencing Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Analysis
| Item | Function in Off-Target Studies | Example/Notes |
|---|---|---|
| High-Fidelity Cas9 Nuclease | Minimizes spurious cleavage. Essential for clean baseline. | Alt-R S.p. HiFi Cas9 Nuclease V3 |
| Synthetic sgRNA (chemically modified) | Improves stability and reduces immune response; enhances specificity. | TruGuide sgRNA with 2'-O-methyl analogs |
| dsODN Tag (for GUIDE-seq) | Oligonucleotide tag integrated into DSBs for unbiased detection. | GUIDESeq dsODN, 5' phosphorylated |
| High-Fidelity PCR Polymerase | Accurate amplification of genomic loci for sequencing with low error rate. | Q5 Hot-Start Polymerase, KAPA HiFi |
| Illumina-Compatible Indexing Kits | Allows multiplexing of many samples/amplicons for NGS validation. | Nextera XT, IDT for Illumina kits |
| Positive Control sgRNA | Known off-target profile to validate experimental and analytical pipelines. | e.g., VEGFA site 3 (well-characterized) |
| CRISPResso2 / Cas-Analyzer | Bioinformatics software for precise quantification of indels from NGS data. | Open-source, web-based or CLI. |
Integrated Framework for CRISPR vs. RNAi Analysis
A direct comparison in functional genomics requires applying these methods to both technologies.
Comparative Off-Target Analysis Workflow
Comparative Off-Target Analysis: CRISPR vs. RNAi
Decision Matrix for Technology Selection
Table 4: Off-Target Considerations for Functional Genomics
| Parameter | CRISPR Knockout | RNAi Knockdown | Implications for Choice |
|---|---|---|---|
| Primary Off-Target Mechanism | DNA mismatch tolerance | Seed-sequence miRNA-like effects | CRISPR: DNA-level validation needed. RNAi: Transcriptome-wide screen needed. |
| Validation Gold Standard | GUIDE-seq / Amp-Seq | Genome-wide RNA-seq | CRISPR costs are locus-focused; RNAi requires full transcriptomics. |
| Typical False Positive Rate (Phenotype) | Lower (complete KO) | Higher (partial KD + transcriptome dysregulation) | CRISPR phenotypes more directly linked to target gene. |
| Mitigation Strategy | High-fidelity Cas9 variants, truncated gRNAs | siRNA chemical modification, pooling | Both allow engineering for specificity. |
| Therapeutic Specificity Barrier | High (permanent edit) | Medium (transient effect) | Drives extreme rigor in CRISPR off-target profiling for clinical apps. |
A rigorous, multi-layered approach combining advanced computational prediction with empirical, sequencing-based validation is non-negotiable for accurately defining the off-target landscape of genome-editing tools. In the context of CRISPR knockout versus RNAi for functional genomics, this systematic approach reveals that while CRISPR offers a more permanent and direct route to loss-of-function, its potential for permanent genomic damage demands exhaustive off-target screening. RNAi, though transient, introduces widespread transcriptional noise. Therefore, the choice between them hinges not only on the desired perturbation but on the acceptable off-target profile for the specific research or therapeutic question, underscored by the methodological rigor outlined herein.
Optimizing Knockout Efficiency (CRISPR) and Knockdown Potency (RNAi)
CRISPR-mediated gene knockout and RNA interference (RNAi)-mediated gene knockdown represent two foundational pillars for loss-of-function studies in functional genomics and drug target validation. The choice between these technologies hinges on the specific research question, the required depth of perturbation, and the experimental system. CRISPR-Cas9 generates permanent, complete loss-of-function alleles via double-strand breaks and error-prone repair, making it ideal for definitive gene knockout. In contrast, RNAi achieves transient, partial reduction of gene expression through mRNA degradation or translational inhibition, suitable for studying essential genes or achieving graded knockdowns. This guide details strategies for optimizing the efficiency and potency of each platform.
Optimizing CRISPR-Cas9 Knockout Efficiency
Core Principle: Knockout efficiency depends on the generation of insertions or deletions (indels) that disrupt the coding frame. Optimization targets sgRNA design, delivery, and cellular repair pathways.
Critical Experimental Protocol: sgRNA Validation & Delivery
Goal: Achieve high-efficiency indel formation in target cell population. Protocol Steps:
- sgRNA Design: Use algorithms (e.g., from Broad Institute, Chop-Chop) to select 2-3 sgRNAs per target. Prioritize exons near the 5' end of the coding sequence, avoiding regions with high sequence homology to prevent off-target effects.
- Cloning: Clone sgRNA sequences into a plasmid expressing both the sgRNA and Cas9 (e.g., lentiCRISPRv2). Include a selectable marker (e.g., puromycin resistance).
- Delivery: Transfect or transduce target cells. For hard-to-transfect cells, use nucleofection or high-titer lentiviral particles.
- Selection: Apply appropriate selection pressure (e.g., puromycin) for 48-72 hours post-delivery.
- Validation: Harvest genomic DNA from the pooled population 5-7 days post-selection. Amplify the target region by PCR and analyze indel frequency via T7 Endonuclease I assay or next-generation sequencing (NGS).
- Clonal Isolation: For homogeneous knockout, single-cell clone the population and screen clones by sequencing to identify bi-allelic disruptions.
Key Optimization Parameters
- sgRNA Efficacy: Use validated design tools and empirical testing of multiple guides.
- Cas9 Activity: Choose the appropriate nuclease (SpCas9, HiFi Cas9 for reduced off-targets).
- Delivery Efficiency: Optimize transfection/transduction protocols for your cell type.
- Repair Pathway Bias: Use small molecule inhibitors (e.g., SCR7 to inhibit NHEJ, promote HDR for knock-ins) or Alt-R HDR Enhancer to bias repair.
Optimizing RNAi Knockdown Potency
Core Principle: Knockdown potency is the degree of mRNA reduction and is influenced by siRNA/shRNA design, efficient delivery, and minimizing off-target effects.
Critical Experimental Protocol: siRNA/shRNA Validation
Goal: Achieve potent, specific, and durable reduction of target mRNA. Protocol Steps:
- Design: Select 3-4 siRNAs per target from validated libraries (e.g., Dharmacon ON-TARGETplus, Qiagen) or design shRNAs using algorithms (e.g., RNAi Codex).
- Delivery (for siRNA): Reverse transfect cells with siRNA using a lipid-based transfection reagent optimized for your cell line. A typical dose is 10-50 nM.
- Delivery (for shRNA): Transduce cells with lentiviral vectors encoding shRNA and a selectable marker.
- Timing: Harvest RNA and protein 48-72 hours post-siRNA transfection or 5-7 days post-shRNA transduction/selection.
- Validation: Quantify mRNA levels via qRT-PCR using TaqMan assays. Confirm protein reduction by western blot. Always include a non-targeting control (NTC) and a positive control (e.g., GAPDH or POLR2A siRNA).
- Dose-Response: Perform an siRNA titration (e.g., 1-50 nM) to establish the concentration for maximal specific knockdown.
Key Optimization Parameters
- Oligo Design: Use chemically modified siRNAs (e.g., 2'-OMe) to enhance stability and reduce immunogenicity.
- Delivery Agent: Screen multiple transfection reagents for highest efficiency and lowest cytotoxicity.
- Controls: Include multiple negative controls and use pooled siRNAs to mitigate off-target effects.
- Validation: Always use orthogonal methods (mRNA + protein) to confirm knockdown.
Quantitative Comparison of Key Metrics
Table 1: Core Performance Characteristics of CRISPR Knockout vs. RNAi Knockdown
| Metric | CRISPR-Cas9 Knockout | RNAi (siRNA/shRNA) Knockdown |
|---|---|---|
| Mechanism | Permanent DNA disruption, indels | Transient mRNA degradation/translational block |
| Efficiency/Potency | High (can achieve >80% indels in pooled pop.) | Variable (typically 70-95% mRNA reduction) |
| Onset of Effect | Rapid (DNA cut in hours), but phenotype may require protein turnover. | Rapid (mRNA reduction within 24h, protein loss by 48-72h). |
| Duration of Effect | Stable, heritable. | Transient (siRNA: 5-7 days; shRNA: can be stable with integration). |
| Primary Pitfalls | Off-target editing, cutting efficiency, phenotypic compensation. | Off-target transcriptional effects, incomplete knockdown, seed-based artifacts. |
| Ideal Application | Essential gene analysis, complete null phenotype, long-term studies, generating stable cell lines. | Studying essential genes where null is lethal, dose-response studies, acute inhibition, in vivo delivery. |
Table 2: Reagent and Tool Optimization Table
| Tool/Reagent | Role in CRISPR Optimization | Role in RNAi Optimization |
|---|---|---|
| High-Efficiency Cas9 | Increases overall editing rates (e.g., eSpCas9). | Not Applicable. |
| Chemically Modified sgRNAs (e.g., Alt-R CRISPR-Cas9 sgRNA) | Enhances stability and reduces immune response in primary cells. | Not Applicable. |
| HDR Enhancers (e.g., Alt-R HDR Donor & Enhancer) | Increases precise editing rates for knock-ins. | Not Applicable. |
| NHEJ Inhibitors (e.g., SCR7) | Can increase ratio of indels to perfect repair. | Not Applicable. |
| Chemically Modified siRNAs (e.g., ON-TARGETplus) | Not Applicable. | Reduces off-target effects and immunostimulation. |
| Lipidoid Nanoparticles (LNPs) | For in vivo delivery of RNP complexes. | For in vivo delivery of siRNA (e.g., Patisiran). |
| Robust Positive Control siRNA (e.g., POLR2A) | Not Applicable. | Essential for protocol and system validation. |
| T7 Endonuclease I / NGS | Critical for quantifying indel efficiency. | Not Applicable (use qPCR/NGS for mRNA). |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Importance |
|---|---|
| Validated sgRNA Libraries (e.g., Brunello, GeCKO v2) | Pre-designed, sequence-verified libraries for genome-wide screens; ensure high on-target activity. |
| HiFi Cas9 Variant | Engineered nuclease with significantly reduced off-target cleavage while maintaining high on-target efficiency. |
| Ribonucleoprotein (RNP) Complexes | Pre-formed Cas9 protein + sgRNA; reduces off-targets, enables rapid editing without delivery of DNA. |
| ON-TARGETplus siRNA Pools | Smart-pool design with chemical modifications to maximize on-target knockdown and minimize off-target effects. |
| Lipofectamine CRISPRMAX / RNAiMAX | Transfection reagents specifically optimized for high-efficiency delivery of CRISPR components or siRNA with low cytotoxicity. |
| TaqMan Gene Expression Assays | Gold-standard for precise, specific quantification of mRNA knockdown levels post-RNAi. |
| Guide-it Genotype Confirmation Kit | Streamlined PCR-to-sequencing workflow for identifying and characterizing CRISPR-edited clones. |
| Control shRNA Lentiviral Particles (Non-Targeting) | Critical for establishing baseline in shRNA experiments, accounting for viral transduction and selection effects. |
Methodological & Pathway Visualizations
Title: CRISPR Knockout Experimental Workflow
Title: RNAi Knockdown Experimental Workflow
Title: CRISPR vs RNAi Selection Logic
Mitigating Adaptive Responses and Phenotypic Escapes in Long-Term Experiments
Within functional genomics research, the choice between CRISPR-Cas9-mediated knockout and RNA interference (RNAi)-mediated knockdown is pivotal for long-term phenotypic studies. A persistent challenge is the emergence of adaptive cellular responses and phenotypic escapes that can obscure experimental results. This guide details strategies to mitigate these confounding effects, ensuring robust and interpretable data in extended experimental timelines.
The core thesis of modern functional genomics often hinges on selecting the optimal loss-of-function technique. CRISPR-Cas9 creates permanent, complete gene knockouts by inducing double-strand breaks, while RNAi achieves transient, partial knockdowns through transcript degradation. In long-term experiments, each approach presents unique pathways for phenotypic escape: CRISPR-edited populations may select for in-frame edits or activate DNA damage response pathways, whereas RNAi-treated cells can exhibit compensatory gene expression or saturation of the RNAi machinery. This guide addresses these mechanisms and provides protocols for their mitigation.
Mechanisms of Adaptive Escape
For CRISPR-Cas9 Knockout Experiments
- Genetic Heterogeneity and Selection: A polyclonal edited population contains a spectrum of indels. Over time, selective pressure can favor subpopulations with in-frame mutations or heterozygous edits that confer a survival advantage, leading to a diluted phenotypic readout.
- p53-Mediated DNA Damage Response: Chronic DNA break induction can activate p53, leading to cell cycle arrest or apoptosis, selectively enriching for p53-deficient clones and introducing a confounding genotype.
- Transcriptional Adaptation: Loss of a gene product can trigger compensatory upregulation of related genes or paralogs, masking the true phenotypic consequence of the initial knockout.
For RNAi Knockout Experiments
- Knockdown Efficiency Decay: Transient transfection or dilution of viral vectors over cell divisions leads to a gradual restoration of target gene expression.
- Target Sequence Mutation: Rare mutations in the siRNA/shRNA target sequence can abolish binding, allowing escapee clones to proliferate.
- Saturation of the Endogenous RNAi Pathway: High, sustained levels of shRNA can compete for and overload exportin-5 and RISC machinery, causing global miRNA dysregulation and cytotoxic off-target effects.
- Compensatory Transcriptional Networks: Cells may activate feedback loops or alternative signaling pathways to bypass the requirement for the knocked-down gene.
Quantitative Comparison of Escape Mechanisms
The following table summarizes the frequency and timelines of key adaptive responses observed in long-term culture studies.
Table 1: Prevalence and Onset of Adaptive Responses in Long-Term Experiments
| Escape Mechanism | Primary Technique | Typical Onset (Weeks Post-Initiation) | Estimated Frequency in Polyclonal Pools | Key Driver(s) |
|---|---|---|---|---|
| Selection for In-Frame Edits | CRISPR Knockout | 4-8 | 10-40% of surviving population | Selective pressure |
| p53 Pathway Adaptation | CRISPR Knockout | 2-6 | 5-20% (cell-type dependent) | Chronic DNA damage |
| Transcriptional Compensation | CRISPR Knockout | 3-10 | Common, difficult to quantify | Network robustness |
| Knockdown Efficiency Decay | RNAi | 1-4 (transient); 4-12 (stable) | Near-ubiquitous without selection | Transient delivery, promoter silencing |
| Target Site Mutation | RNAi | 6-16 | 1-10% under strong selection | RNAi-mediated mutagenesis or rare genomic mutation |
| RISC/MicroRNA Pathway Saturation | RNAi (shRNA) | 1-3 | Significant with high-copy, potent shRNAs | High shRNA expression |
Experimental Protocols for Mitigation
Protocol 1: Designing and Validating a Redundant CRISPR gRNA Strategy
Purpose: To minimize escape via genetic heterogeneity and selection. Materials: CRISPR design software (e.g., CHOPCHOP, Benchling), oligos for gRNA cloning, lentiviral packaging system, next-generation sequencing (NGS) platform.
- Design: Identify 3-5 independent gRNAs targeting early exons of the gene of interest. Prioritize gRNAs with high on-target and low off-target scores.
- Clone: Clone each gRNA individually and as a pooled combination into your Cas9-expression vector (e.g., lentiCRISPRv2).
- Transduce & Select: Transduce target cells at a low MOI (<0.3) to ensure single-copy integration. Apply appropriate selection (e.g., puromycin) for 5-7 days.
- Validate Editing: At 1-week post-selection, harvest genomic DNA from the polyclonal pool. Perform PCR amplification of all target loci and subject to NGS. Calculate frameshift efficiency for each gRNA individually and the pool.
- Long-Term Culture: Culture the validated polyclonal pool for the experiment duration. Re-assess editing efficiency monthly via NGS to monitor for selection of in-frame alleles.
Protocol 2: Monitoring p53 Response in CRISPR-Edited Cells
Purpose: To detect and control for confounding p53 pathway activation. Materials: Antibodies for p53 and p21 (Western), flow cytometer, apoptosis detection kit (e.g., Annexin V).
- Establish Controls: Include a non-targeting gRNA control and a gRNA targeting a known essential gene as positive controls.
- Temporal Sampling: Harvest cells at 72 hours, 1 week, and 2 weeks post-editing/selection.
- Analysis:
- Western Blot: Analyze lysates for p53 and p21 protein levels. Sustained elevation indicates DNA damage response activation.
- Flow Cytometry: Assess cell cycle profile (propidium iodide staining) for G1 arrest. Perform Annexin V/propidium iodide staining to quantify apoptosis.
- Interpretation: If the specific gene knockout consistently activates p53 compared to non-targeting control, consider using a p53 inhibitor (e.g., small molecule) transiently during editing or switching to an alternative cell line with compromised p53 signaling, acknowledging this as a limitation.
Protocol 3: Ensuring Sustained Knockdown in RNAi Experiments
Purpose: To prevent phenotypic escape due to loss of knockdown. Materials: Multiple distinct shRNA constructs, inducible expression system (e.g., Tet-On), qRT-PCR reagents.
- Use Inducible Systems: Employ a doxycycline-inducible shRNA system. Maintain cells in continuous doxycycline throughout the experiment. Include an "uninduced" control population from the same pool.
- Employ Combinatorial RNAi: Clone 2-3 distinct shRNAs targeting the same gene into a single vector or co-transduce with separate vectors.
- Continuous Selection: Maintain antibiotic selection (e.g., puromycin) for the entire experiment if using constitutively expressed shRNAs.
- Rigorous QC: Perform qRT-PCR and/or Western blot analysis on samples collected in parallel with functional assays, not just at the experiment's start. This confirms sustained target suppression at the time of phenotypic measurement.
Visualizing Mitigation Strategies
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Mitigating Adaptive Responses
| Reagent / Solution | Primary Function | Example / Note |
|---|---|---|
| Lentiviral gRNA/shRNA Vectors | Stable genomic integration of genetic perturbation elements. | lentiCRISPRv2 (CRISPR), pLKO.1-TRC (shRNA), Tet-pLKO-puro (Inducible shRNA). |
| Next-Generation Sequencing (NGS) Kits | Quantifying editing efficiency, detecting heterogeneity, and tracking clone evolution. | Illumina MiSeq for amplicon sequencing; services for CRISPResso2 analysis. |
| p53/p21 Antibodies | Monitoring DNA damage response activation in CRISPR experiments via Western Blot. | Phospho-specific p53 (Ser15) antibodies add sensitivity. |
| Doxycycline (or Analog) | Induction and maintenance of shRNA expression in Tet-On systems for sustained knockdown. | Use at calibrated minimal effective concentration to reduce pleiotropic effects. |
| Puromycin / Other Selection Antibiotics | Maintaining constant selective pressure on cells harboring perturbation constructs. | Titer carefully; continuous low-dose is often sufficient for maintenance. |
| Validated Silencer Select siRNAs or shRNAs | High-quality, pre-validated RNAi reagents with published efficiency scores to reduce initial variability. | Using 2-3 distinct sequences per gene is recommended. |
| cDNA Rescue Construct | Expressing a wild-type or modified (RNAi-resistant) version of the target gene to confirm on-target effects. | Critical control to distinguish on-target from adaptive/off-target phenotypes. |
| Cell Viability & Apoptosis Assay Kits | Quantifying cytotoxic responses that may lead to selective pressure and escape. | e.g., Annexin V/Propidium Iodide for flow cytometry; Real-time cell analysis (e.g., xCELLigence). |
| Single-Cell Cloning Dilution Media | Isolating monoclonal populations from polyclonal edits to reduce heterogeneity. | Often contains high serum or conditioned media to support low-density growth. |
Successful long-term functional genomics experiments require preemptive planning for adaptive responses. CRISPR knockout studies demand careful monitoring of genetic heterogeneity and DNA damage responses, while RNAi experiments necessitate strategies for sustained suppression. Implementing the redundant design, continuous validation, and parallel approach protocols outlined here will significantly reduce phenotypic escapes. Ultimately, the most robust conclusions are drawn when key phenotypes are reproduced using both CRISPR and RNAi, as concordant results are less likely to be artifacts of either technique's specific escape pathways.
In functional genomics, the choice between CRISPR-Cas9 knockout and RNA interference (RNAi) knockdown is foundational. A core thesis posits that CRISPR offers superior specificity and permanent gene silencing, while RNAi allows for tunable, transient knockdown but suffers from off-target effects. However, the experimental validity of this thesis hinges entirely on technical efficiency. Low efficiency—whether from poor delivery, suboptimal expression, or inadequate reagent QC—can obscure true biological outcomes, leading to false conclusions in comparative studies. This guide provides a systematic, technical framework for diagnosing and resolving these critical bottlenecks.
Delivery: Navigating Cellular Barriers
Delivery remains the primary rate-limiting step. The modality must be matched to the cell type and the technology (CRISPR’s large Cas9 ribonucleoprotein vs. RNAi’s smaller siRNA/shRNA).
Quantitative Comparison of Delivery Methods
Table 1: Efficiency and Toxicity Profiles of Common Delivery Methods
| Method | Typical Efficiency (Hard-to-Transfect Cells) | Typical Toxicity | Best Suited For | Key Challenge |
|---|---|---|---|---|
| Lipofection (CRISPR Plasmid) | 10-40% | Moderate | Standard cell lines (HEK293, HeLa) | Cytoplasmic degradation, endosomal trapping |
| Lipofection (siRNA) | 50-90% | Low-Moderate | Most adherent cell lines | Nuclease degradation, RISC competition |
| Electroporation (RNP/siRNA) | 70-95% | High | Primary cells, immune cells, neurons | Cell mortality, require optimization |
| Lentiviral Transduction (shRNA) | >90% (with selection) | Low (post-infection) | Long-term knockdown, in vivo studies | Insertional mutagenesis, variable copy number |
| Adeno-associated Virus (AAV) | Varies by serotype | Very Low | In vivo delivery, non-dividing cells | Limited cargo capacity (~4.7 kb) excludes SpCas9 |
| Microinjection (RNP) | >95% | Low (per cell) | Zygotes, single-cell studies | Low throughput, highly specialized |
Detailed Protocol: Electroporation for Primary T-Cells (CRISPR RNP)
This protocol optimizes delivery while minimizing toxicity.
- Isolate and Activate: Isolate primary human T-cells and activate for 48 hours using CD3/CD28 Dynabeads in IL-2 supplemented media.
- Prepare RNP Complex: For a single reaction, complex 30 pmol of purified S.p. Cas9 protein with 30 pmol of synthetic sgRNA (resuspended in nuclease-free duplex buffer) by incubating at 25°C for 10 minutes.
- Wash and Resuspend: Harvest activated T-cells, remove beads, wash twice in PBS, and resuspend at 1x10^7 cells/mL in pre-warmed, serum-free electroporation buffer (e.g., P3 primary cell solution).
- Electroporation: Mix 100 µL cell suspension (1x10^6 cells) with 10 µL pre-complexed RNP. Transfer to a 100 µL electroporation cuvette. Use a nucleofector device with the recommended pulse code (e.g., EH-115 for human T-cells).
- Immediate Recovery: Immediately post-pulse, add 500 µL of pre-warmed, serum-rich media to the cuvette. Transfer cells to a pre-coated (e.g., RetroNectin) 24-well plate containing complete media + IL-2.
- Analysis: Assess editing efficiency via flow cytometry (using a surrogate reporter) or T7E1 assay at 72 hours post-electroporation.
Diagram 1: CRISPR RNP Delivery Workflow for Primary T-Cells
Expression & Design: Maximizing Target Engagement
Efficient expression of CRISPR or RNAi components is non-negotiable.
For CRISPR:
- Promoter Selection: The U6 promoter is standard for sgRNA. For Cas9, use cell-type-appropriate promoters (EF1α, CAG for broad expression; synapsin for neurons).
- sgRNA Design: Use validated algorithms (CRISPick, CHOPCHOP) and prioritize on-target scores. Always include a positive control sgRNA (e.g., targeting AAVS1 or HPRT1).
- Expression Kinetics: Plasmid-based expression is slow (24-72h). For rapid analysis, use pre-complexed RNP.
For RNAi:
- shRNA vs. siRNA: Chemically synthesized siRNA offers immediate action but is transient. Lentiviral shRNA allows stable integration but requires careful titration to avoid interferon response.
- Design Rules: Follow Tuschl rules. Use pooled siRNA to mitigate off-targets. Validate with multiple independent sequences per gene.
Detailed Protocol: Validating sgRNAIn VitroCleavage
A critical QC step before cellular experiments.
- PCR Amplification: Generate a 500-1000 bp genomic target fragment containing the sgRNA site from human genomic DNA using high-fidelity PCR.
- Purify Substrate: Gel-purify the PCR product and quantify accurately.
- In Vitro Cleavage Assay: Set up a 20 µL reaction: 200 ng PCR product, 100 nM purified Cas9 nuclease, 120 nM sgRNA (complexed as in delivery protocol), 1x Cas9 reaction buffer. Incubate at 37°C for 1 hour.
- Analysis: Run the product on a 2% agarose gel. Compare to a non-targeting sgRNA control. Efficient cleavage yields two lower molecular weight bands. Quantify cleavage efficiency using gel analysis software (% cleaved = intensity of cut bands / total intensity).
Reagent QC: The Foundation of Reproducibility
Poor reagent quality is a silent killer of efficiency.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Critical Reagents for CRISPR/RNAi Experiments and Their Function
| Reagent / Material | Function & Importance | Key QC Checkpoint |
|---|---|---|
| Nuclease-Free Water | Solvent for oligos, buffers. RNase contamination destroys RNA reagents. | Certificates of Analysis (CoA) for RNase testing; use aliquots. |
| Synthetic sgRNA (Chemically Modified) | Directly guides Cas9 to DNA target. Stability affects RNP half-life. | HPLC/MS purification, PAGE analysis for integrity, endotoxin testing. |
| Purified Cas9 Protein | The effector enzyme. Purity impacts specificity and cellular toxicity. | SDS-PAGE for single band >95%, functional in vitro cleavage assay, absence of endotoxins. |
| Positive Control siRNA/sgRNA | Essential for troubleshooting delivery/expression systems. | Target a ubiquitously expressed gene (GAPDH, PPIB, HPRT1). Validate in your cell line. |
| Lipofectamine 3000 | Common lipid-based transfection reagent for plasmids/siRNA. | Perform a dose-response with a GFP reporter plasmid to optimize ratio for each cell line. |
| CRISPR-Cas9 Plasmid (all-in-one) | Expresses Cas9 and sgRNA from same vector. Convenient for screening. | Sequence verify sgRNA cloning, confirm promoter activity via transfection & Western blot for Cas9. |
| Lentiviral Packaging Mix (2nd/3rd Gen) | For producing shRNA or CRISPR lentivirus. Safety and titer are critical. | Use a functional titer assay (e.g., p24 ELISA and puromycin kill curve or GFP% analysis). |
| RISC-Free siRNA (or shRNA) | For RNAi negative controls. Chemically modified to prevent loading into RISC. | Verify lack of phenotype in genome-wide transcriptomic analysis if possible. |
Diagram 2: Logical Troubleshooting Pathway for Low Efficiency
Rigorous troubleshooting of delivery, expression, and reagent QC is not mere technique—it is the prerequisite for meaningful scientific comparison. In the context of CRISPR knockout vs. RNAi, only experiments executed at high efficiency can accurately reveal the distinct advantages and limitations of each technology: the consistent, complete loss-of-function from CRISPR versus the graded, transient suppression from RNAi. By adhering to the stringent protocols and validations outlined here, researchers can ensure their data robustly informs the functional genomics thesis, driving reliable discovery in basic research and drug development.
Within functional genomics, the choice between CRISPR-mediated knockout (CRISPRko) and RNA interference (RNAi) remains pivotal. While CRISPRko induces permanent DNA breaks leading to frameshift mutations, RNAi achieves transient mRNA degradation. This whitepaper details the critical experimental controls—scrambled, non-targeting, and essential genes—required to validate findings and mitigate false positives/negatives in both systems, thereby ensuring robust data interpretation.
The Role of Controls in CRISPRko vs. RNAi Experiments
Controls are essential to account for non-specific effects inherent to each technology.
- CRISPRko: Controls must rule out off-target DNA cleavage and phenotypic effects from the transfection/nucleofection process or the Cas9 protein itself.
- RNAi: Controls must account for sequence-independent immune responses (e.g., interferon activation) and saturation of the endogenous RNAi machinery (seed-based off-targets).
The table below summarizes key differential considerations:
Table 1: Control Considerations for CRISPRko vs. RNAi
| Control Aspect | CRISPRko | RNAi |
|---|---|---|
| Primary Artifact Source | Off-target DNA double-strand breaks. | Seed region-mediated off-target mRNA knockdown. |
| Delivery Confound | Cas9/gRNA RNP or plasmid toxicity. | Lipofection/electroporation stress; siRNA concentration-dependent immune activation. |
| Timeline for Effects | Permanent; requires single-cell cloning for clean analysis. | Transient; peak knockdown typically 48-96h post-transfection. |
| Optimal Non-Targeting Control | gRNA with no genomic target (validated by sequencing). | Scrambled siRNA with no significant homology to any transcriptome (validated by microarray/NGS). |
| Essential Gene Application | Confirms editing efficiency and delivery; cell death expected. | Confirms knockdown efficiency and delivery; strong growth impairment expected. |
Design and Implementation of Critical Controls
A. Scrambled & Non-Targeting Controls
These are "negative" controls designed to mimic the experimental intervention without targeting any biologically relevant sequence.
Protocol: Designing and Validating a Non-Targeting gRNA for CRISPRko
- Design: Use a bioinformatics tool (e.g., CHOPCHOP, Broad Institute GPP Portal) to design a 20nt guide sequence lacking homology to the target genome. BLAST the sequence against the relevant genome assembly.
- Specificity Check: Ensure the guide has at least 3 mismatches to any other genomic site, especially in the seed region (positions 1-12 proximal to PAM).
- Cloning: Clone into your chosen gRNA expression vector (e.g., lentiCRISPRv2, pX459).
- Validation: Transduce/transfect cells and perform targeted deep sequencing (e.g., Illumina MiSeq) across the top in silico predicted off-target sites (typically up to 10 sites) to confirm absence of indels. Compare to parental cell line.
Protocol: Designing and Validating a Scrambled siRNA for RNAi
- Sequence Generation: Randomly scramble the nucleotide sequence of your active siRNA. Maintain similar GC content (~30-50%).
- BLAST Validation: Perform a stringent nucleotide BLAST (blastn) against the RefSeq RNA database to ensure no significant homology (>16-17nt contiguous identity) to any non-target transcript.
- Functional Test: Transfect the scrambled siRNA (at the same concentration as experimental siRNAs) into cells and assay for global gene expression changes via microarray or RNA-seq 24-48h post-transfection. A valid control should show minimal (e.g., <50 genes differentially expressed) non-specific perturbation.
B. Essential Gene Controls
These are "positive" controls confirming the technical efficacy of the knockout or knockdown process.
Protocol: Using Essential Gene Controls (e.g., POLR2A, RPL7A)
- Gene Selection: Choose a gene essential for cell survival/proliferation (e.g., POLR2A for RNA polymerase, RPL7A for ribosomes, CASP3 for apoptosis). Public databases (e.g., DepMap) provide validated essential genes by cell line.
- Reagent Delivery: For CRISPRko, transfect cells with a validated, high-efficiency gRNA targeting the essential gene. For RNAi, transfert with a validated siRNA (from publications or reputable vendors).
- Phenotypic Readout: Monitor viability 3-7 days (CRISPRko) or 3-5 days (RNAi) post-treatment using a robust assay (e.g., ATP-based luminescence, high-content imaging for confluence).
- Efficiency Validation: Confirm on-target activity:
- For CRISPRko: Perform T7 Endonuclease I assay or tracking of indels by decomposition (TIDE) analysis on genomic DNA at 48-72h.
- For RNAi: Perform qRT-PCR on mRNA extracted 48-72h post-transfection.
- Interpretation: An effective experiment must show significant cell death or growth inhibition (>70% reduction vs. non-targeting control) for the essential gene control. Failure indicates issues with delivery, reagent potency, or assay sensitivity.
Experimental Workflow & Data Integration
The following diagram illustrates the integration of these controls into a standard functional genomics screening or validation workflow.
Diagram Title: Control Integration in Functional Genomics Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Implementing Robust Controls
| Reagent / Solution | Function in Control Experiments | Example Product/Catalog |
|---|---|---|
| Validated Non-Targeting gRNA | Negative control for CRISPRko; packaged in lentiviral or all-in-one plasmid format. | Dharmacon Edit-R Non-targeting Control; Sigma-Aldrich MISSION CRISPR Control Plasmid. |
| Validated Scrambled siRNA | Negative control for RNAi; chemically modified to reduce immune stimulation. | Dharmacon ON-TARGETplus Non-targeting Control; Ambion Silencer Select Negative Control. |
| Validated Essential Gene gRNA/siRNA | Positive control to confirm system functionality. | Horizon Discovery Essential Gene Control Kit (e.g., PLK1); Sigma-Aldrich MISSION siRNA POLR2A. |
| Cas9 Nuclease (WT) | Required for CRISPRko; used as protein (RNP) or expressed from plasmid. | Integrated DNA Technologies Alt-R S.p. Cas9 Nuclease; ToolGen Recombinant Cas9 Protein. |
| Lipofectamine Transfection Reagent | For delivery of siRNA/gRNA plasmids; requires optimization for each cell line. | Thermo Fisher Lipofectamine RNAiMAX (for siRNA); Lipofectamine 3000 (for plasmid). |
| Viability/Proliferation Assay Kit | Quantify essential gene control effect and target phenotype. | Promega CellTiter-Glo (ATP-based); Roche xCELLigence (Real-time impedance). |
| Genomic DNA Extraction Kit | Prepare samples for CRISPRko editing efficiency validation (TIDE, NGS). | Qiagen DNeasy Blood & Tissue Kit; Zymo Research Quick-DNA Miniprep Kit. |
| qRT-PCR Kit (One-Step) | Validate RNAi knockdown efficiency for essential and target genes. | Bio-Rad iScript One-Step RT-PCR; Thermo Fisher TaqMan RNA-to-CT. |
| T7 Endonuclease I | Enzyme for detecting CRISPR-induced indels at genomic target sites. | New England Biolabs T7 Endonuclease I (M0302S). |
The strategic implementation of scrambled/non-targeting and essential gene controls is non-negotiable for rigorous functional genomics. These controls directly address the distinct mechanistic artifacts of CRISPRko and RNAi, enabling researchers to attribute phenotypes accurately to on-target gene loss. As the field progresses towards higher-throughput and more complex phenotypic screens, these best practices form the bedrock of reliable, interpretable data crucial for both basic research and drug discovery pipelines.
Head-to-Head Validation: A Comparative Framework for Confident Gene Function Assignment
Direct Comparison of Phenotypic Concordance and Discordance
Within the broader thesis comparing CRISPR-mediated knockout and RNAi-mediated knockdown for functional genomics, the analysis of phenotypic concordance and discordance is paramount. RNAi reduces gene expression post-transcriptionally, often incompletely and with off-target effects, while CRISPR/Cas9 generates permanent, complete gene knockouts. Phenotypic outcomes from these distinct perturbations can align (concordance) or diverge (discordance). Understanding the drivers of discordance is critical for interpreting functional genomics screens, validating targets, and advancing drug discovery. This guide provides a technical framework for their direct comparison.
A. Mechanism of Action:
- CRISPR Knockout: Indels cause frameshifts/nonsense mutations, leading to complete loss of functional protein. Effects are permanent and observable over multiple cell generations.
- RNAi Knockdown: Degrades mRNA, leading to partial reduction (typically 70-90%) of target protein. Effects are transient and subject to compensatory cellular mechanisms.
B. Off-Target Effects:
- RNAi: Well-documented seed-sequence-based off-target mRNA degradation. Requires careful control design (e.g., rescue experiments, multiple sh/siRNAs).
- CRISPR: DNA-level off-target cleavage at sites with sequence homology. Mitigated by high-fidelity Cas9 variants, optimized guide RNA design, and careful validation.
C. Temporal Dynamics & Compensation:
- Acute RNAi knockdown may elicit different cellular responses than a stable CRISPR knockout, where genetic compensation or network rewiring can occur.
Table 1: Comparison of CRISPR-KO and RNAi Methodological Attributes
| Attribute | CRISPR Knockout | RNAi Knockout | Notes |
|---|---|---|---|
| Target Molecule | Genomic DNA | mRNA | Fundamental difference |
| Efficacy (Protein Reduction) | ~100% (complete knockout) | 70-95% (knockdown) | Key driver of discordance |
| Phenotype Onset | Delayed (requires cell division/depletion) | Rapid (hours to days) | |
| Duration of Effect | Permanent, stable | Transient (days to a week) | |
| Primary Off-Target Risk | DNA cleavage at homologous sites | mRNA degradation via seed region | |
| Typical Controls | Non-targeting guide, rescue with cDNA | Non-targeting siRNA, multiple target sequences | |
| Screening Readiness | Excellent for positive selection; clone outgrowth can bias negative selection. | Suitable for acute phenotypes in pooled/arrayed formats. |
Table 2: Reported Phenotypic Concordance Rates from Comparative Studies
| Study (Key Finding) | System | Reported Concordance Rate | Major Source of Discordance Identified |
|---|---|---|---|
| Evers et al., 2016 | Cancer cell line proliferation | ~60-80% | Off-target effects (RNAi), incomplete knockdown |
| Shalem et al., 2014 | Melanoma resistance to vemurafenib | High for top hits; divergence in secondary hits. | RNAi false positives from off-targets |
| Morgens et al., 2016 | Genome-wide viability screens | ~70% concordance for essential genes. | Hypomorphic RNAi phenotypes vs. null CRISPR phenotypes |
Experimental Protocols for Direct Comparison
Protocol 1: Head-to-Head Phenotypic Screening Validation
- Objective: Compare hit identification from a genome-wide CRISPR knockout library vs. an RNAi library for a specific phenotype (e.g., drug resistance).
- Method:
- Cell Line: Use a genetically stable, rapidly dividing cell line (e.g., A375 melanoma, K562 leukemic).
- Perturbation: In parallel, transduce with genome-wide CRISPR knockout (e.g., Brunello) library and genome-wide shRNA (e.g., TRC) library at low MOI to ensure single integrations.
- Selection & Phenotype Induction: Apply puromycin selection. Split cells and apply phenotypic pressure (e.g., drug treatment) vs. vehicle control (DMSO).
- Harvest & Sequencing: Harvest genomic DNA from pre-selection and post-selection populations. Amplify integrated guide/shRNA sequences via PCR.
- Analysis: Sequence amplicons. Calculate enrichment/depletion scores (e.g., MAGeCK, RSA). Rank hits. Compare gene lists from both screens using rank-rank hypergeometric overlap analysis.
Protocol 2: Single-Gene Validation of Discordant Hits
- Objective: Mechanistically dissect why a specific gene shows a phenotype with one technology but not the other.
- Method:
- CRISPR-KO Validation: Design 2-3 independent sgRNAs. Perform clonal isolation via limiting dilution. Validate knockout by Sanger sequencing (TIDE analysis) and Western blot.
- RNAi Validation: Transferd with 2-3 independent siRNA sequences or transduce with 2-3 distinct shRNA lentiviral constructs. Confirm knockdown 72h post-transfection by qRT-PCR and Western blot.
- Phenotype Assay: Perform the relevant phenotypic assay (e.g., Incucyte proliferation, apoptosis assay, migration) on both KO and KD cells alongside appropriate controls.
- Rescue Experiment (Critical): For discordant phenotypes, reintroduce a CRISPR/Cas9-resistant cDNA (silent mutations in PAM/protospacer) into the CRISPR-KO clone. Test if phenotype is rescued. If phenotype appears only with RNAi, rescue suggests on-target effect; if not, suggests off-target.
Visualizations
Diagram Title: Phenotype Concordance and Discordance Decision Workflow
Diagram Title: CRISPR-KO vs RNAi Mechanistic Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Comparative Studies
| Reagent / Solution | Function in Comparison Studies | Key Considerations |
|---|---|---|
| Genome-wide CRISPR Knockout Library (e.g., Brunello, TorontoKO) | Enables loss-of-function screening. Provides single sgRNA per gene with high on-target scores. | Use libraries with minimal sgRNA redundancy. Employ high-fidelity Cas9 cell lines. |
| Genome-wide shRNA Library (e.g., TRC, Decipher) | Enables knockdown-based screening. | Use libraries with multiple shRNAs per gene to control for off-targets. |
| High-Fidelity Cas9 (e.g., SpCas9-HF1, eSpCas9) | Reduces DNA off-target cleavage in CRISPR experiments. | Critical for improving specificity vs. RNAi. |
| Non-Targeting Control sgRNA/shRNA | Controls for non-specific effects of transduction/transfection and RISC/DNA cleavage machinery. | Sequence should be validated for no genomic target. |
| CRISPR-Resistant cDNA Construct | Expresses target gene with silent mutations in the sgRNA target site. The gold standard for rescue validation. | Confirms on-target effects and dissects discordance. |
| Next-Generation Sequencing Kit (for amplicon seq) | Quantifies guide/shRNA abundance from pooled screens. | Essential for calculating enrichment/depletion statistics. |
| TIDE (Tracking of Indels by Decomposition) Analysis Tool | Quickly assesses CRISPR editing efficiency from Sanger traces without clonal isolation. | Enables rapid validation of knockout. |
| Phenotypic Assay Kits (e.g., Caspase-3, MTS, Real-Time ATP) | Quantifies the specific cellular phenotype under investigation. | Must be optimized for the time window post-perturbation (acute vs. chronic). |
In functional genomics research, the choice between CRISPR-Cas9-mediated knockout and RNA interference (RNAi) for gene silencing is fundamental. While RNAi achieves transient knockdown via mRNA degradation, CRISPR-Cas9 creates permanent genetic modifications. A critical step following any perturbation is rigorous validation of the on-target effect. This guide details the application of three core techniques—Western Blot, quantitative PCR (qPCR), and Next-Generation Sequencing (NGS)—to confirm target protein ablation or mRNA knockdown, directly informing the interpretation of downstream phenotypic assays within a CRISPR vs. RNAi thesis.
Western Blot for Protein-Level Validation
Western Blot is the gold standard for confirming changes in target protein abundance, crucial for distinguishing between CRISPR knockout (complete absence) and RNAi knockdown (partial reduction).
Detailed Protocol:
- Cell Lysis: Harvest cells 48-72h post-transfection/transduction. Lyse in RIPA buffer supplemented with protease/phosphatase inhibitors. Centrifuge at 14,000 x g for 15 min at 4°C.
- Protein Quantification: Use a BCA assay. Load equal amounts (20-40 µg) of protein per lane onto a 4-20% gradient SDS-PAGE gel.
- Electrophoresis & Transfer: Run gel at 120V for 90 min. Transfer to PVDF membrane using a wet-transfer system at 100V for 70 min on ice.
- Blocking & Incubation: Block membrane with 5% non-fat milk in TBST for 1h. Incubate with primary antibody (diluted in blocking buffer) overnight at 4°C. Use a loading control antibody (e.g., GAPDH, β-Actin) simultaneously.
- Detection: Incubate with HRP-conjugated secondary antibody for 1h at RT. Develop with chemiluminescent substrate and image.
Key Data Interpretation: CRISPR knockouts should show a complete absence of the target protein band. RNAi treatments typically show a quantifiable reduction.
Table 1: Quantitative Outcomes from Western Blot Analysis
| Perturbation Method | Expected Protein Reduction | Key Indicator | Typical Timeline for Analysis |
|---|---|---|---|
| CRISPR-Cas9 KO | 95-100% | Absence of band | 72-96h post-transduction |
| RNAi (siRNA/shRNA) | 50-90% | Band intensity reduction | 48-72h post-transfection |
qPCR for mRNA-Level Validation
qPCR quantifies changes in target mRNA transcript levels, essential for verifying RNAi efficacy and assessing CRISPR knockouts that may trigger nonsense-mediated decay (NMD).
Detailed Protocol:
- RNA Extraction: Use TRIzol or column-based kits. Include DNase I treatment to eliminate genomic DNA.
- cDNA Synthesis: Use 1 µg of total RNA with random hexamers or oligo-dT primers in a reverse transcription reaction.
- qPCR Setup: Prepare reactions in triplicate using SYBR Green or TaqMan chemistry. Use a 20 µL reaction volume.
- Primer Design: Design exon-spanning primers for RNAi validation. For CRISPR validation, design primers flanking the cut site (to detect indel-induced amplicon size changes) and a separate set for transcript quantification.
- Cycle Conditions: 95°C for 3 min, followed by 40 cycles of 95°C for 10 sec and 60°C for 30 sec.
- Data Analysis: Calculate ∆∆Ct values normalized to housekeeping genes (e.g., ACTB, GAPDH).
Table 2: qPCR Validation Signatures
| Method | Primer Design Focus | Expected mRNA Change | Notes |
|---|---|---|---|
| RNAi | Targeting transcript | 60-95% reduction | Confirms on-target knockdown efficiency. |
| CRISPR-Cas9 KO | Flanking cut site & transcript | Variable; often >70% reduction | Transcript decay via NMD; flanking primers may show altered amplification efficiency. |
NGS for Genomic-Level Confirmation
NGS provides the most definitive proof of on-target editing by CRISPR-Cas9, precisely characterizing indel spectra and allele frequencies. It is less common for RNAi validation.
Detailed Protocol for Amplicon Sequencing:
- Genomic DNA Extraction: Use a column-based kit 3-7 days post-editing.
- PCR Amplification: Design primers (~150-200 bp flanking the target site) with overhangs for Illumina indexing. Use a high-fidelity polymerase.
- Library Preparation: Clean amplicons and perform a limited-cycle indexing PCR. Use a dual-indexing strategy to minimize cross-talk.
- Sequencing: Pool libraries and sequence on a MiSeq or similar platform (2x250 bp or 2x300 bp).
- Data Analysis: Use CRISPR-specific variant callers (e.g., CRISPResso2, ICE Synthego) to quantify indel percentage and visualize allele distributions.
Table 3: NGS Analysis Metrics for CRISPR Validation
| Metric | Definition | Acceptable Benchmark for Clonal Validation |
|---|---|---|
| % Indel Frequency | Percentage of reads with indels at cut site. | >70% in pooled populations; 100% in clones. |
| % Wild-Type | Percentage of unmodified reads. | As low as possible. |
| Predominant Indel Size | Most common insertion/deletion length. | Guides phenotypic prediction (frameshift vs. in-frame). |
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Function & Application |
|---|---|
| High-Fidelity Polymerase (e.g., Q5, KAPA) | Error-free amplification for NGS amplicon prep and cloning. |
| CRISPR-Cas9 RNP Complex | Direct delivery of Cas9 protein and gRNA for reduced off-targets. |
| Validated siRNA Pools | Pre-designed, pooled siRNAs for robust RNAi knockdown. |
| HRP-conjugated Secondary Antibodies | Essential for chemiluminescent detection in Western Blot. |
| SYBR Green qPCR Master Mix | Cost-effective, sensitive detection of mRNA levels. |
| Dual-Indexing Adapter Kits (Illumina) | For multiplexed NGS of CRISPR-edited amplicons. |
| NGS Analysis Software (CRISPResso2) | Specialized tool for quantifying CRISPR editing outcomes. |
Experimental Workflow & Logical Relationships
Decision Flow for Validating Gene Perturbations
Multi-Method Validation Workflow for CRISPR/RNAi
Within functional genomics research, the choice between CRISPR-Cas9-mediated knockout (KO) and RNA interference (RNAi) for gene silencing is fundamental. While both aim to elucidate gene function by reducing target expression, their mechanisms of action are distinct, leading to inherent differences in their propensity for false-positive and false-negative results. This guide provides a technical assessment of these limitations, emphasizing that the experimental context dictates which tool's artifacts are most consequential. Accurate interpretation of functional genomics screens and validation experiments requires a rigorous understanding of these context-dependent pitfalls.
Core Mechanisms & Inherent Artifact Profiles
RNAi operates at the transcript level via the RNA-induced silencing complex (RISC), leading to targeted mRNA degradation or translational repression. Its key artifacts stem from:
- Off-target effects: Partial sequence complementarity between the siRNA/shRNA and non-target mRNAs, leading to unintended transcript knockdown and false positives.
- Seed-region mediated miRNA-like effects: The siRNA "seed sequence" (nucleotides 2-8) can repress multiple transcripts with complementary 3'UTRs, confounding phenotypes.
- Incomplete knockdown: Residual target protein (often 70-90% reduction) can be sufficient for function, leading to false negatives, especially for stable proteins or genes with high expression.
- Competitive saturation of endogenous miRNA machinery: High concentrations of shRNA can disrupt natural microRNA regulation, causing indirect effects.
CRISPR-Cas9 Knockout creates permanent, DNA-level disruptions via double-strand breaks (DSBs) repaired by error-prone non-homologous end joining (NHEJ). Its key artifacts stem from:
- Off-target effects: Cas9 nuclease activity at genomic loci with sequence homology to the guide RNA (gRNA), particularly in regions with limited mismatches in the PAM-distal seed region, causing unintended mutations and false positives.
- On-target, non-functional edits: In-frame indels or nucleotide substitutions that do not disrupt the protein's function, leading to false negatives (allelic heterogeneity).
- p53-mediated cell state perturbations: DSBs can activate the p53 pathway, inducing apoptosis or cell cycle arrest in sensitive cell lines, creating a phenotype independent of the target gene's function.
- Genetic compensation: Some KO models trigger transcriptional adaptation via related genes, masking loss-of-function phenotypes (false negatives).
The following diagram illustrates the primary mechanisms and artifact pathways for each technology.
Quantitative Comparison of Artifact Rates
The table below summarizes typical quantitative measures for key limitations, based on recent literature. These values are highly dependent on experimental design parameters (e.g., gRNA/siRNA design, delivery method, cell type).
Table 1: Comparative Rates of Artifacts in RNAi vs. CRISPR-KO Screens
| Artifact Metric | RNAi (siRNA/shRNA) | CRISPR-Cas9 Knockout | Notes & Contextual Dependence |
|---|---|---|---|
| On-Target Efficacy | 70-95% mRNA knockdown | >90% frameshift rate (varies by locus) | CRISPR efficacy is binary (KO/no KO); RNAi leaves residual protein. |
| Off-Target Rate (Transcriptome-wide) | High (up to 10s-100s of genes per siRNA) | Lower, but significant with poor gRNAs | RNAi off-targets are predictable by seed sequence; CRISPR off-targets are sequence-dependent. |
| False Negative Rate | Higher for stable proteins, redundant genes, high-abundance transcripts | Higher for genes where in-frame edits are tolerated, or with genetic compensation | RNAi FNs from incomplete KD; CRISPR FNs from non-disruptive edits. |
| False Positive Rate (from tool-specific artifacts) | High in screens sensitive to miRNA-like seed effects | High in screens sensitive to DNA damage response (e.g., viability in p53-wt cells) | Context is critical: p53 artifacts irrelevant for in vitro biochemical assays. |
| Phenotype Concordance Between Tools | ~60-70% for core essential genes | Higher self-correlation between multiple gRNAs | Discordance often reveals tool-specific artifacts or biology (e.g., dosage sensitivity). |
Detailed Experimental Protocols for Artifact Mitigation
Protocol 4.1: Validating RNAi On-Target Specificity & Overcoming False Negatives
This protocol uses rescue with an RNAi-resistant cDNA to confirm phenotype specificity.
- Design: Identify the target siRNA sequence. Use silent mutagenesis (codon optimization) to design a cDNA construct of the target gene that is resistant to the siRNA (mutates the siRNA binding site without altering the amino acid sequence).
- Cloning: Clone the wild-type and RNAi-resistant cDNAs into a mammalian expression vector with a selectable marker (e.g., puromycin) and an independent fluorescent tag (e.g., GFP).
- Cell Line Generation: Transfect the target cell line with the empty vector, WT-cDNA, or resistant-cDNA constructs. Select with appropriate antibiotic for 5-7 days to create stable polyclonal pools.
- Knockdown & Assay: Reverse-transfect siRNA targeting the endogenous gene (and a non-targeting control) into each stable cell pool. Incubate for 72-96 hours (allowing protein turnover).
- Validation & Analysis:
- Western Blot: Confirm knockdown of endogenous protein (absent in WT-cDNA pool) and expression of the resistant transgene.
- Phenotypic Assay: Perform the functional assay (e.g., viability, migration, reporter activity). A phenotype observed in the empty-vector and WT-cDNA pools that is rescued specifically in the resistant-cDNA pool confirms on-target activity. Failure to rescue suggests an off-target artifact.
Protocol 4.2: Validating CRISPR-Cas9 Knockout Specificity & Assessing False Negatives from In-Frame Edits
This protocol uses deep sequencing and single-cell cloning to assess editing heterogeneity.
- Transduction & Editing: Transduce cells with lentivirus delivering Cas9 and a specific gRNA. Select with appropriate antibiotics (e.g., blasticidin for Cas9, puromycin for gRNA).
- Bulk Population Analysis: After 7-10 days, extract genomic DNA from the polyclonal population. PCR-amplify the target region (~300-400bp around cut site) using barcoded primers.
- Next-Generation Sequencing (NGS): Purify and quantify the amplicon library. Sequence on a MiSeq or similar platform to high coverage (>5000x).
- Bioinformatic Analysis: Use tools like CRISPResso2 to quantify the spectrum of indels. Calculate the percentage of in-frame mutations (indels of multiples of 3 nucleotides). A high percentage (>30%) suggests a risk of false negatives in a bulk population assay.
- Single-Cell Cloning: To isolate a pure knockout, perform limiting dilution of the polyclonal population to generate single-cell clones. Expand clones for 2-3 weeks.
- Genotype & Phenotype Correlation: Screen clones by PCR and Sanger sequencing to identify those with biallelic frameshift mutations. In parallel, perform the functional assay on multiple independent KO clones and non-edited control clones. Correlate the genotype (presence of frameshift) with the phenotype. Lack of phenotype in a verified frameshift clone may indicate genetic compensation or false-positive p53 artifacts, requiring further validation (e.g., rescue with cDNA, p53 knockout).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Mitigating False Results in Functional Genomics
| Reagent Category | Specific Example/Product | Function in Artifact Assessment |
|---|---|---|
| Control RNAs | Non-targeting siRNA (e.g., Silencer Select Negative Control) / Non-targeting gRNA (e.g., targeting safe-harbor locus like AAVS1) | Baseline control for delivery and non-specific immune responses. |
| Rescue Constructs | RNAi-resistant ORF clones (e.g., from Horizon Discovery's Rescue-MATE) / CRISPR-resistant "CRISPR-Proof" cDNAs | Gold-standard validation of on-target phenotype for both technologies. |
| Improved Nuclease Variants | High-fidelity SpCas9 (SpCas9-HF1, eSpCas9) / HypaCas9 | Reduce CRISPR off-target cleavage by weakening non-specific DNA interactions. |
| p53 Pathway Modulators | p53 inhibitor (e.g., Pifithrin-α) / Isogenic p53-/- cell lines | Identify false-positive viability hits in CRISPR screens due to DNA damage toxicity. |
| Multiplicity Tools | siRNA pools (4-6 per gene) / gRNA libraries (3-6 per gene) | Mitigate false positives/negatives from a single RNA guide; phenotype consistency across multiple guides implies on-target effect. |
| NGS-Based Off-Target Profiling Kits | GUIDE-seq / CIRCLE-seq / SITE-seq kits | Empirically map genome-wide off-target sites for a given gRNA or siRNA seed region. |
| Phenotypic Assay Controls | Essential gene positive controls (e.g., PLK1, RPA3) / Non-essential gene negative controls (e.g., AAVS1, ROSA26) | Benchmark screen performance and Z'-factors to assess assay robustness independent of tool. |
Contextual Decision Framework: Choosing the Right Tool
The choice between RNAi and CRISPR-KO is not universal but depends on the biological question, system, and readout. The following workflow diagram provides a logical framework for this decision.
Conclusion: A rigorous functional genomics study acknowledges that both CRISPR-KO and RNAi are prone to distinct, context-specific false results. RNAi's strengths lie in probing dosage-sensitive phenomena and acute knockdowns but are confounded by off-target transcript repression. CRISPR-KO provides definitive, permanent ablation but introduces DNA-damage artifacts and allelic heterogeneity. The most reliable conclusions are drawn when results converge across both technologies, or when robust rescue and clonal validation experiments are performed. The choice of tool must be a deliberate, hypothesis-driven decision, not a default setting.
Cost, Time, and Scalability Analysis for High-Throughput Functional Genomics
Within the pivotal debate of CRISPR knockout versus RNAi for functional genomics research, selecting the optimal technology extends beyond biological mechanism to encompass practical constraints of cost, time, and scalability. This guide provides a quantitative and methodological framework for researchers and drug development professionals to analyze these operational dimensions, enabling data-driven platform selection for high-throughput screens.
Core Technology Comparison: Operational Metrics
The fundamental differences in the mechanism of action between RNAi (transient knockdown) and CRISPR-Cas9 (permanent knockout) propagate into distinct experimental timelines, cost structures, and scalability profiles.
Table 1: High-Level Operational Comparison: CRISPR-KO vs. RNAi
| Parameter | CRISPR-Cas9 Knockout | RNA Interference (RNAi) |
|---|---|---|
| Primary Mechanism | Permanent gene disruption via DSBs and NHEJ. | Transient mRNA degradation via siRNA/shRNA. |
| Typical Screening Format | Arrayed or pooled lentiviral sgRNA libraries. | Arrayed synthetic siRNA or pooled shRNA libraries. |
| Experimental Timeline (From design to data) | Longer (weeks-months). Requires clonal expansion or deep sequencing analysis. | Shorter (days-weeks). Phenotype assessed post-transfection. |
| Reagent Cost per Genome Screen | Higher (sgRNA library, Cas9, sequencing). | Lower (synthetic siRNA pools). |
| Scalability for Genome-Wide | Excellent for pooled formats; arrayed is resource-intensive. | Very high for arrayed synthetic siRNA. |
| Major Cost/Time Drivers | Viral production, NGS validation, clonal outgrowth. | Reagent synthesis, repeat transfections for kinetics. |
Detailed Cost Breakdown Analysis
Costs are segmented into startup (fixed) and per-screen (variable) components. Current market data (2023-2024) indicates the following structure.
Table 2: Cost Analysis for a Genome-Wide Human Screen (~20,000 genes)
| Cost Category | CRISPR-Cas9 Pooled Screen | RNAi (Arrayed Synthetic siRNA) |
|---|---|---|
| Startup / Capital | Stable Cas9 cell line generation (~$2k-$5k in reagents/time). | Optimization of transfection protocol (minimal). |
| Library Reagents | Pooled sgRNA lentiviral library: $3,000 - $7,000. | Arrayed siRNA library (4 oligos/gene): $15,000 - $25,000. |
| Delivery / Infection | Viral packaging & titering: $1,500 - $3,000. | Transfection reagent: $2,000 - $4,000 for screen. |
| Cell Culture & Consumables | Media, antibiotics, plates: ~$2,000. | Media, plates: ~$3,000 (more plates needed). |
| Phenotypic Assay | Assay-dependent (e.g., CellTiter-Glo): $1,500 - $5,000. | Assay-dependent: $1,500 - $5,000. |
| Readout & Validation | Major cost driver. NGS of sgRNA abundance: $4,000 - $10,000. Hit validation via indel detection (T7E1, NGS): $1,000 - $5,000. | RT-qPCR for mRNA knockdown: $1,000 - $3,000. Hit confirmation with alternative siRNAs: $500 - $2,000. |
| Estimated Total Direct Reagent Cost | $12,000 - $35,000 | $22,000 - $42,000 |
| Key Cost Insight | Lower library cost but significant NGS readout expense. Cost-effective for many genes/pool. | Higher upfront library cost, but lower per-screen readout cost. Efficient for targeted or sub-genomic sets. |
Experimental Timeline & Workflow
Time is a critical, often overlooked, resource. The workflows differ substantially.
Table 3: Comparative Timeline for a Functional Genomics Screen
| Phase | CRISPR-Cas9 Pooled Screen (Weeks) | RNAi Arrayed Screen (Weeks) |
|---|---|---|
| 1. Project Planning & Design | 1-2 | 1-2 |
| 2. Library Acquisition & QC | 2-4 (viral production & titering) | 1-2 (siRNA re-arraying) |
| 3. Pilot Optimization | 2-3 (MOI, selection efficiency) | 1-2 (transfection efficiency, toxicity) |
| 4. Main Screen Execution | 2-3 (infection, selection, phenotypic challenge) | 1-2 (reverse transfection, phenotypic assay) |
| 5. Sample Prep for Readout | 1-2 (genomic DNA extraction, PCR for NGS) | Immediate (direct lysate for luminescence/imaging) |
| 6. Data Acquisition & Analysis | 2-4 (NGS run, bioinformatic analysis) | 1 (plate reader data processing) |
| 7. Primary Hit Validation | 3-6 (single-clone isolation, validation of knockout) | 1-2 (repeat with alternative oligos, RT-qPCR) |
| Total Estimated Timeline | 13 - 26 weeks | 7 - 13 weeks |
Detailed Methodologies
Protocol: Pooled CRISPR-Cas9 Screen with NGS Readout
Objective: To identify genes essential for cell viability under a specific condition. Materials:
- Cas9-expressing cell line.
- Pooled lentiviral sgRNA library (e.g., Brunello, 4 sgRNAs/gene).
- Polybrene (8 µg/mL) to enhance infection.
- Puromycin for selection.
- PCR primers for amplifying sgRNA inserts from genomic DNA.
- Next-generation sequencing platform.
Procedure:
- Viral Transduction: Plate cells and transduce with library virus at an MOI~0.3 to ensure single integration. Include a non-transduced control.
- Selection: 24h post-transduction, add puromycin (dose determined by kill curve) for 5-7 days.
- Phenotypic Challenge: Split selected cell population. Maintain one arm under control conditions and the other under experimental challenge (e.g., drug treatment). Culture for 14-21 population doublings.
- Genomic DNA (gDNA) Harvest: Extract gDNA from a minimum of 200 cells per sgRNA at T0 (post-selection) and from both control and experimental arms at endpoint. Use a column-based maxi-prep kit.
- sgRNA Amplification & Sequencing: Perform a two-step PCR. PCR1: Amplify sgRNA cassette from ~100 µg gDNA per sample using library-specific primers. PCR2: Add Illumina adaptors and sample barcodes. Purify amplicons.
- NGS & Analysis: Pool samples and sequence on an Illumina HiSeq/NovaSeq (75bp single-end). Align reads to the reference library. Use MAGeCK or similar tool to compare sgRNA abundance between conditions and rank essential genes.
Protocol: Arrayed RNAi Screen with Luminescent Readout
Objective: To identify genes that modulate a specific signaling pathway using a luciferase reporter. Materials:
- Arrayed siRNA library (384-well format, 20-40 nM final).
- Reverse transfection reagent (e.g., Lipofectamine RNAiMAX).
- Luciferase reporter cell line.
- CellTiter-Glo for viability assessment.
- One-Glo or Dual-Glo Luciferase assay system.
- Automated liquid handler & plate reader.
Procedure:
- Plate Library: Using an acoustic dispenser or pin tool, transfer siRNA stocks to assay plates.
- Reverse Transfection: Dilute transfection reagent in Opti-MEM, add to assay plates, incubate 15 min. Seed reporter cells directly into the lipid-siRNA complex.
- Incubation: Incubate for 72-96 hours to allow knockdown.
- Stimulation & Assay: Stimulate pathway if required (e.g., add ligand). Perform Dual-Glo assay: first, measure firefly luciferase (pathway signal); then, add Stop & Glo reagent to quench firefly and activate Renilla luciferase (normalization control).
- Viability Assay: In parallel plate, perform CellTiter-Glo at endpoint.
- Data Analysis: Normalize firefly to Renilla signal. Calculate Z-scores. Apply viability cut-off (e.g., >70% cell health). Prioritize hits that significantly alter the reporter signal without causing toxicity.
Scalability Considerations
Scalability involves throughput, reagent logistics, and data management.
Table 4: Scalability Matrix
| Dimension | CRISPR-KO (Pooled) | RNAi (Arrayed) |
|---|---|---|
| Theoretical Gene Throughput | Extremely High (entire genome in one experiment). | High, but limited by plate density (e.g., 10k genes in 30x 384wp). |
| Reagent Logistics | Simple post-pooling (one viral mixture). Complex pre-pooling library management. | Complex (managing thousands of discrete siRNA wells). |
| Phenotypic Flexibility | Limited to assays compatible with mixed cell populations (proliferation, FACS sorting, NGS-based reporters). | Very High. Compatible with any plate-based assay (imaging, luminescence, FRET). |
| Hit Deconvolution | Requires NGS and bioinformatics; false positives from multiple sgRNAs per gene needed. | Directly known from well position. |
| Automation Friendliness | Low for pooled steps, high for NGS prep. | Very High. Ideal for full robotic automation. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 5: Key Reagents for High-Throughput Functional Genomics
| Reagent / Solution | Function in Screen | Example Product/Supplier |
|---|---|---|
| Arrayed siRNA Library | Pre-dispensed, gene-specific siRNA pools for arrayed screens. | Dharmacon siGENOME, Qiagen HP GenomeWide. |
| Pooled sgRNA Lentiviral Library | Barcoded viral vectors for pooled CRISPR screens. | Broad Institute GPP (Brunello), Addgene pooled libraries. |
| Reverse Transfection Reagent | Enables high-throughput lipid-mediated siRNA delivery in plate format. | Invitrogen Lipofectamine RNAiMAX, Mirus Bio TransIT-X2. |
| Cas9 Stable Cell Line | Provides constitutive Cas9 expression, simplifying pooled screens. | Synthego Knockout Cell Lines, generate via lentivirus + blasticidin selection. |
| Viral Packaging System | Produces lentivirus for sgRNA library delivery. | psPAX2 & pMD2.G packaging plasmids, Mirus Bio TransIT-Lenti. |
| NGS Library Prep Kit for CRISPR | Streamlines amplification and barcoding of sgRNAs from gDNA. | Illumina Nextera XT, New England Biolabs NEBNext Ultra II. |
| Cell Viability Assay (Luminescent) | Measures cytotoxicity for hit triage in arrayed screens. | Promega CellTiter-Glo. |
| Genomic DNA Extraction Kit (Maxi) | High-yield, high-quality gDNA prep from pooled cell pellets. | Qiagen Blood & Cell Culture DNA Maxi Kit. |
| Bioinformatics Analysis Software | Statistical analysis of screen hits from NGS or plate data. | MAGeCK (CRISPR), CellHTS2 (RNAi), proprietary solutions (Genedata). |
Visualizations
Diagram 1 Title: CRISPR vs RNAi screening workflow comparison.
Diagram 2 Title: Major cost drivers in functional genomics screens.
Diagram 3 Title: Scalability vs phenotypic flexibility trade-off.
The choice between CRISPR knockout and RNAi for high-throughput functional genomics is context-dependent. CRISPR pooled screens offer superior biological certainty of loss-of-function and are highly scalable in terms of gene number, but at a higher per-screen cost and longer timeline, with constraints on phenotypic assays. RNAi arrayed screens provide faster, more flexible phenotypic interrogation with direct hit identification, making them ideal for focused or pathway-specific studies, though library costs are higher for genome-wide coverage. The optimal strategy aligns the technology's operational profile—detailed in cost, time, and scalability dimensions—with the specific biological question, assay requirements, and resource constraints of the research program.
Functional genomics aims to elucidate gene function through perturbation and observation. Two predominant technologies dominate this landscape: CRISPR-Cas9-mediated knockout (KO) and RNA interference (RNAi)-mediated knockdown (KD). Each method possesses distinct mechanistic strengths and limitations, creating a complementary framework for rigorous gene validation.
CRISPR-Cas9 creates permanent, DNA-level deletions or insertions, resulting in complete gene knockout. This is ideal for studying essential genes or long-term phenotypic consequences but may be confounded by clonal variation, off-target editing, and adaptive compensatory mechanisms.
RNAi (including siRNA and shRNA) reduces gene expression post-transcriptionally via targeted mRNA degradation. It allows for tunable, transient suppression and is suitable for studying essential genes where complete knockout is lethal. However, it is prone to incomplete knockdown, off-target effects via miRNA-like seed region binding, and transient effects.
The integrative tandem approach leverages these complementary profiles. Using both technologies sequentially or in parallel provides a powerful orthogonal validation strategy, where phenotypes observed with both methods can be attributed to on-target gene modulation with high confidence, mitigating the limitations inherent to each technique alone.
Core Comparative Data: CRISPR vs. RNAi
Table 1: Mechanistic and Performance Comparison of CRISPR-KO and RNAi-KD
| Feature | CRISPR-Cas9 Knockout | RNA Interference (si/shRNA) |
|---|---|---|
| Molecular Target | Genomic DNA | mRNA |
| Mechanism | Double-strand break → indels via NHEJ/MMEJ | RISC-mediated mRNA cleavage or translational repression |
| Effect on Protein | Complete, permanent loss | Partial, reversible reduction |
| Typical Efficiency | High (often >70% indels in pool) | Variable (70-95% mRNA reduction) |
| Key Technical Duration | Permanent, stable | Transient (siRNA: days; shRNA: weeks) |
| Major Advantage | Complete loss-of-function; epigenetic editing possible | Tunable knockdown; applicable to essential genes |
| Primary Limitation | Off-target DNA cleavage; clonal variation | Off-target transcript effects (seed-based); incomplete knockdown |
| Optimal Use Case | Definitive loss-of-function; synthetic lethality screens | Dose-dependent studies; essential gene analysis |
Table 2: Phenotypic Concordance Metrics from Tandem Studies (Representative Data) Data synthesized from recent tandem screening publications (2023-2024).
| Gene Class | % Genes with Phenotype in CRISPR-KO Only | % Genes with Phenotype in RNAi-KD Only | % Genes with Concordant Phenotype (Both Platforms) | Key Inference |
|---|---|---|---|---|
| Essential Genes | 5-15% | 25-40% | 45-60% | RNAi reveals hypomorphic phenotypes; CRISPR defines core essentials. |
| Kinases/Enzymes | 20-30% | 10-20% | 50-60% | CRISPR identifies non-catalytic roles; RNAi sensitive to protein turnover. |
| Transcription Factors | 40-50% | 10-20% | 30-40% | High CRISPR-specific rate suggests strong adaptation to complete KO. |
Tandem Experimental Strategy: Sequential and Parallel Workflows
The tandem confirmation strategy can be deployed in two primary modes: Sequential Validation and Parallel Orthogonal Screening.
Sequential Validation Workflow
This is the most common approach, where a hit from a primary large-scale screen (e.g., a CRISPR library screen) is validated using the orthogonal technology (e.g., RNAi).
Protocol 1: CRISPR Hit Validation with RNAi Objective: Confirm a phenotypic hit from a CRISPR screen using siRNA-mediated knockdown.
- Hit Identification: Select top candidate genes from CRISPR pooled screen data (based on guide RNA enrichment/depletion statistics).
- Design Orthogonal Probes: Design 3-4 independent siRNA duplexes per target gene using updated algorithms (e.g., from Horizon Discovery, Qiagen). Ensure no overlap with the CRISPR target genomic region.
- Transfection: Plate cells in 96-well format. Transfect with individual siRNAs using a lipid-based transfection reagent (e.g., Lipofectamine RNAiMAX). Include non-targeting siRNA (negative control) and a siRNA targeting an essential gene (positive control for phenotypic effect).
- Knockdown Efficiency QC: 48 hours post-transfection, harvest a plate for RNA extraction. Perform qRT-PCR to confirm mRNA knockdown (>70% recommended).
- Phenotype Re-assessment: At the assay-appropriate time (e.g., 72-96h for proliferation), measure the phenotype (e.g., viability, luminescence, imaging) in the siRNA-treated cells.
- Analysis: A hit is considered robustly validated if ≥2 independent siRNAs recapitulate the direction and significance of the CRISPR phenotype.
Parallel Orthogonal Screening
A more resource-intensive but comprehensive approach where both perturbations are applied in parallel from the outset.
Protocol 2: Parallel CRISPR and RNAi Pooled Screening Objective: Identify high-confidence hits with inherent orthogonal validation.
- Library Design & Pooling:
- CRISPR arm: Use a genome-wide sgRNA library (e.g., Brunello, TorontoKO).
- RNAi arm: Use a genome-wide shRNA library (e.g., TRC, in a miR-E backbone) or a parallel siRNA screening library.
- Viral Transduction (for CRISPR/shRNA): Perform separate transductions for the CRISPR and shRNA libraries at a low MOI (<0.3) to ensure single integration. Use appropriate selection (puromycin for CRISPR, puromycin/blasticidin for shRNA).
- Screening & Sequencing: Conduct the phenotypic screen (e.g., dropout in the presence of a drug). Harvest genomic DNA (for CRISPR gDNA and shRNA barcode PCR) at T0 and Tfinal.
- Bioinformatic Integration: Analyze screen data separately using standard pipelines (MAGeCK for CRISPR, RIGER for shRNA). Overlap hits from both screens using rank-based methods (e.g., rank-product). Genes that score significantly in both independent screens represent the highest-confidence hits.
Essential Pathways and Workflows
Tandem Confirmation Decision Workflow
Mechanistic Pathways of CRISPR and RNAi
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Tandem CRISPR/RNAi Studies
| Reagent Category | Specific Example/Supplier | Key Function in Tandem Approach |
|---|---|---|
| CRISPR Nucleases | Alt-R S.p. HiFi Cas9 (IDT), Edit-R Cas9 (Horizon) | High-fidelity Cas9 variants to minimize off-target editing during initial KO. |
| sgRNA Design/ Synthesis | Custom Alt-R crRNAs (IDT), Synthego CRISPR kits | Chemically modified guides for enhanced stability and specificity. |
| RNAi Triggers | ON-TARGETplus siRNA (Horizon), Silencer Select (Ambion) | Pre-designed, validated siRNA pools with reduced seed-based off-target effects. |
| Delivery Reagents | Lipofectamine CRISPRMAX (for RNP), RNAiMAX (for siRNA) | Optimized lipid nanoparticles for efficient, low-toxicity delivery of different payloads. |
| Libraries | Brunello CRISPR KO Lib (Addgene), DECIPHER shRNA Lib (Horizon) | Genome-wide, validated pooled libraries for parallel screening. |
| Validation Assays | T7 Endonuclease I / ICE Analysis (CRISPR), RT-qPCR kits (RNAi) | Tools to quantify editing efficiency (indel %) and knockdown efficacy (% mRNA remaining). |
| Cell Lines | Stable Cas9-expressing lines (e.g., HEK293T-Cas9), Reporter lines | Engineered lines to streamline workflows; reporter lines for rapid phenotype readout. |
| Next-Gen Sequencing Kits | Illumina Nextera XT, NGS for guide/barcode sequencing | For quantifying guide/barcode abundance in pooled screening outcomes. |
Conclusion
Choosing between CRISPR knockout and RNAi is not a matter of identifying a universally superior technology, but of strategically matching the tool to the specific biological question, experimental timeline, and required certainty level. CRISPR knockout offers definitive, permanent gene ablation ideal for essential gene identification and modeling loss-of-function mutations, while RNAi provides a faster, often more scalable approach for transient knockdowns and initial target prioritization. The future of functional genomics lies in their complementary use—employing RNAi for rapid screening and CRISPR for deep validation—and in the continued evolution of both platforms, such as high-fidelity Cas variants and enhanced RNAi delivery. For drug development, this rigorous, comparative approach is critical for de-risking target identification and building a robust chain of evidence from gene to therapeutic candidate.