CRISPRa vs CRISPRi: A Comprehensive Guide to Transcriptional Control for Researchers
This article provides a detailed scientific comparison of CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) technologies.
CRISPRa vs CRISPRi: A Comprehensive Guide to Transcriptional Control for Researchers
Abstract
This article provides a detailed scientific comparison of CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) technologies. Tailored for researchers, scientists, and drug development professionals, it explores the foundational mechanisms, distinct methodologies, and practical applications of these powerful transcriptional control tools. We cover experimental design, best-practice protocols, common troubleshooting strategies, and head-to-head comparisons of efficacy, specificity, and versatility. The guide concludes by evaluating current validation standards and future implications for functional genomics, therapeutic development, and precision medicine.
Understanding the Core: How CRISPRa and CRISPRi Differ from CRISPR-Cas9
Within the broader thesis of CRISPRa vs CRISPRi explained research, this whitepaper delineates the fundamental mechanistic and operational differences between CRISPR-based transcriptional modulation (CRISPRa/i) and traditional genomic cleavage (CRISPR-Cas9). While CRISPR-Cas9 creates permanent DNA double-strand breaks (DSBs) to knock out genes, CRISPR activation (CRISPRa) and interference (CRISPRi) enable precise, reversible upregulation or downregulation of gene expression without altering the underlying DNA sequence. This guide provides an in-depth technical comparison for researchers and drug development professionals.
Core Mechanisms and Components
CRISPR-Cas9 for Genomic Cleavage
The native Streptococcus pyogenes CRISPR-Cas9 system functions as a programmable nuclease. A guide RNA (gRNA) directs the Cas9 nuclease to a complementary genomic locus, where it induces a DSB. Repair via non-homologous end joining (NHEJ) often results in insertions/deletions (indels) that disrupt the gene.
CRISPRa/i for Transcriptional Perturbation
CRISPRa and CRISPRi repurpose a catalytically "dead" Cas9 (dCas9), which retains DNA-binding ability but lacks nuclease activity. Transcriptional control is achieved by fusing dCas9 to effector domains:
- CRISPRi: dCas9 is fused to transcriptional repressors (e.g., KRAB, Mxi1). The complex binds near the transcription start site (TSS), physically blocking RNA polymerase or recruiting chromatin-condensing machinery to silence gene expression.
- CRISPRa: dCas9 is fused to transcriptional activators (e.g., VP64, p65, Rta). Advanced systems like SunTag or SAM (Synergistic Activation Mediator) recruit multiple activators. The complex binds to promoter or enhancer regions, recruiting co-activators and the transcriptional apparatus to boost gene expression.
Quantitative Comparison of Key Parameters
Table 1: Core Functional Comparison
| Parameter | CRISPR-Cas9 (Cleavage) | CRISPRi (Interference) | CRISPRa (Activation) |
|---|---|---|---|
| Cas9 Form | Wild-type, nuclease-active | Catalytically dead (dCas9) | Catalytically dead (dCas9) |
| Primary Action | Creates DNA double-strand breaks | Blocks transcription initiation | Recruits transcriptional activators |
| Genetic Change | Permanent indels/mutations | Epigenetic/steric, no DNA change | Epigenetic, no DNA change |
| Outcome | Gene knockout (loss-of-function) | Gene knockdown (reduced expression) | Gene overexpression (gain-of-function) |
| Reversibility | Permanent | Reversible (upon dCas9-effector removal) | Reversible (upon dCas9-effector removal) |
| Typical Efficiency | High (70-90% indels) | High (70-95% repression) | Variable (2-20x activation; system-dependent) |
| Key Risk | Off-target cleavages, p53 activation | Off-target binding, potential seed-mediated toxicity | Off-target binding, overexpression toxicity |
Table 2: Applications in Drug Discovery & Functional Genomics
| Application | CRISPR-Cas9 | CRISPRi | CRISPRa |
|---|---|---|---|
| Target Validation | Essential gene knockout studies | Tunable, reversible knockdown | Gain-of-function phenotyping |
| Screening Modality | Knockout screens (negative selection) | Knockdown screens (hypomorphic alleles) | Activation screens (positive selection) |
| Therapeutic Modality | Ex vivo cell therapy (e.g., CAR-T), in vivo gene disruption | Targeting haploinsufficient diseases, metabolic tuning | Upregulating protective genes, therapeutic proteins |
| Modeling Disease | Creating knockout cell/animal models | Modeling partial loss-of-function, dosage sensitivity | Modeling gene overexpression, oncogene activation |
Experimental Protocols
Protocol: Pooled CRISPRi/a Knockdown/Activation Screen
Objective: Identify genes whose suppression (CRISPRi) or overexpression (CRISPRa) confers a selective growth advantage or disadvantage under a specific condition.
- Library Design: Obtain a pooled lentiviral gRNA library targeting the desired gene set (e.g., whole-genome, druggable genome). For CRISPRi, design gRNAs to target regions -50 to +300 bp relative to the TSS. For CRISPRa, target gRNAs to enhancer regions or within -400 to -50 bp upstream of the TSS.
- Virus Production: Co-transfect HEK293T cells with the library plasmid, psPAX2 (packaging), and pMD2.G (VSV-G envelope) plasmids using PEI transfection reagent. Harvest lentiviral supernatant at 48 and 72 hours.
- Cell Infection & Selection: Transduce target cells (e.g., a cancer cell line stably expressing dCas9-KRAB for CRISPRi or dCas9-SunTag for CRISPRa) at a low MOI (~0.3) to ensure single gRNA integration. Select transduced cells with puromycin (2-5 µg/mL) for 7 days.
- Screen Conduct: Split cells into experimental and control arms (e.g., drug treatment vs. DMSO). Maintain cells for 14-21 population doublings, keeping >500x library coverage at all steps.
- Genomic DNA Extraction & NGS: Harvest pellets (~1e7 cells per sample). Extract gDNA (Qiagen Maxi Prep). Amplify integrated gRNA sequences via PCR using indexed primers. Sequence on an Illumina HiSeq.
- Data Analysis: Align sequences to the reference library. Use MAGeCK or pinAPL-Py algorithms to compare gRNA abundance between conditions and identify significantly enriched or depleted genes.
Protocol: Validation of Transcriptional Perturbation (RT-qPCR)
Objective: Quantitatively measure changes in mRNA expression following CRISPRa/i perturbation.
- Cell Transfection/Nucleofection: Deliver dCas9-effector and target-specific gRNA plasmids (or ribonucleoprotein complexes) into cells.
- Incubation: Allow 72-96 hours for effector recruitment and transcriptional modulation.
- RNA Isolation: Lyse cells and isolate total RNA using a column-based kit (e.g., RNeasy, Zymo Research) with on-column DNase I digestion.
- cDNA Synthesis: Synthesize cDNA from 1 µg RNA using a Reverse Transcription kit (e.g., High-Capacity cDNA Reverse Transcription, Applied Biosystems) with random hexamers.
- qPCR: Prepare reactions with SYBR Green master mix, cDNA template, and gene-specific primers. Run in triplicate on a real-time PCR system. Use GAPDH or ACTB as housekeeping controls.
- Analysis: Calculate fold-change using the 2^(-ΔΔCt) method relative to a non-targeting gRNA control.
Diagrams of Core Mechanisms
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPRa/i Research
| Reagent / Material | Supplier Examples | Critical Function |
|---|---|---|
| dCas9-KRAB (for CRISPRi) Plasmid | Addgene (Plasmid #71237), Sigma-Aldrich | Provides the core silencing machinery: dCas9 + Kruppel-associated box repressor. |
| dCas9-VP64 or SunTag System (for CRISPRa) | Addgene (e.g., Plasmid #61423, #60903), Takara Bio | Provides the core activation machinery. SunTag allows recruitement of multiple VP64 units. |
| Synergistic Activation Mediator (SAM) System | Horizon Discovery, Synthego | Three-component system (dCas9-VP64, MS2-gRNA, MS2-p65-HSF1) for robust activation. |
| Pooled Lentiviral gRNA Libraries | Dharmacon (Edit-R), Sigma (Mission), Cellecta | Pre-designed, barcoded libraries for genome-wide or pathway-specific screens. |
| Chemically Modified Synthetic gRNAs | Synthego, IDT | Enhanced stability and binding affinity for improved on-target efficiency and reduced immunogenicity. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene | Essential for producing lentiviral particles to deliver CRISPR components. |
| Puromycin, Blasticidin, or other Selection Agents | Thermo Fisher, Invivogen | Used to select for cells stably expressing dCas9-effector and/or gRNA constructs. |
| Next-Generation Sequencing Kits (for gRNA amplicons) | Illumina (Nextera XT), NEB Next | For quantifying gRNA abundance from genomic DNA in pooled screening. |
| MAGeCK or pinAPL-Py Software | Open Source (Bioinformatics) | Statistical tool for analyzing CRISPR screen data to identify hit genes. |
Within the broader thesis comparing CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), understanding the core recruitment mechanism of transcriptional activators is paramount. CRISPRa, unlike CRISPRi which represses gene expression, is designed to upregulate target genes. This whitepaper details the molecular engineering behind two dominant CRISPRa systems: the VPR fusion and the Synergistic Activation Mediator (SAM) complex. These systems repurpose a catalytically dead Cas9 (dCas9) as a programmable DNA-binding scaffold to recruit transcriptional activation machinery to specific genomic loci, offering powerful tools for functional genomics and therapeutic development.
Core Mechanism: dCas9 as a Programmable Scaffold
The foundation of CRISPRa is dCas9, which lacks endonuclease activity but retains its ability to bind DNA guided by a single guide RNA (sgRNA). This creates a precise, targetable platform. The core innovation lies in fusing or recruiting potent transcriptional activation domains (ADs) to this dCas9-sgRNA complex.
The VPR Fusion System
The VPR system involves the direct fusion of a tripartite activator, VPR, to the C-terminus of dCas9. VPR is a synthetic fusion of three strong ADs: VP64, p65, and Rta.
- VP64: Four tandem copies of the Herpes Simplex Viral Protein 16 (VP16) AD.
- p65: A subunit of the NF-κB complex.
- Rta: A transcriptional activator from Epstein-Barr virus. This fusion creates a highly potent, single-component CRISPRa system where dCas9-VPR is recruited directly by the sgRNA.
The Synergistic Activation Mediator (SAM) System
The SAM system is a more complex, multi-component recruitment strategy. It relies on orthogonal RNA-protein interactions to recruit multiple copies of the p65-HSF1 AD.
- The sgRNA is engineered with two additional RNA aptamers in its tetraloop and stemloop 2 (MS2 and PP7 aptamers).
- These aptamers are bound by matching coat proteins (MCP and PCP), which are fused to the p65-HSF1 AD.
- Simultaneously, the dCas9 protein itself is fused to a weak AD, VP64. This results in the synergistic recruitment of multiple ADs to a single genomic site, leading to robust gene activation.
Quantitative Comparison of CRISPRa Systems
The following table summarizes key quantitative performance metrics for VPR and SAM, highlighting differences critical for experimental design.
Table 1: Performance Comparison of Major CRISPRa Systems
| Feature | dCas9-VPR | dCas9-SAM |
|---|---|---|
| Core Architecture | Single fusion protein (dCas9-VPR). | Multi-component: engineered sgRNA + dCas9-VP64 + MCP-p65-HSF1 + PCP-p65-HSF1. |
| Activation Domains | VP64-p65-Rta fusion. | VP64 (on dCas9) + multiple p65-HSF1 (recruited via RNA aptamers). |
| Typical Fold Activation | 50 - 300x (varies by target gene) | 100 - 1000x (often higher than VPR for many targets) |
| System Size | ~5.7 kb for the dCas9-VPR expression construct. | Larger: ~5.2 kb (dCas9-VP64) + ~2.6 kb (activation helper) + engineered sgRNA. |
| Delivery Complexity | Lower (two components: dCas9-VPR + sgRNA). | Higher (three+ components: dCas9-VP64, helper protein, engineered sgRNA). |
| Background Noise | Generally low. | Can be higher due to leaky expression of helper components. |
| Key Advantage | Simplicity, high activity in a compact format. | Very high activation levels due to synergistic recruitment. |
Detailed Experimental Protocol: CRISPRa Activation Assay
This protocol outlines a standard experiment to test the efficacy of a CRISPRa system (e.g., VPR or SAM) in activating a target gene in cultured mammalian cells.
A. Materials and Reagents
- Cells: HEK293T or relevant cell line.
- Plasmids:
- CRISPRa Expression Plasmid: e.g., pLV-dCas9-VPR or pLV-dCas9-VP64 (for SAM).
- sgRNA Expression Plasmid: For VPR: standard sgRNA backbone. For SAM: sgRNA backbone with MS2 and PP7 aptamers (e.g., pLenti-sgRNA-MS2-PP7).
- SAM Helper Plasmid (if using SAM): Expressing MCP-p65-HSF1 and PCP-p65-HSF1 fusion proteins.
- Reporter Plasmid (Optional): Containing a minimal promoter driving a fluorescent protein (e.g., GFP) downstream of the target sequence.
- Transfection Reagent: e.g., Lipofectamine 3000.
- qPCR Reagents: SYBR Green mix, primers for target gene and housekeeping gene (e.g., GAPDH).
- Flow Cytometry Buffer: PBS with 2% FBS.
B. Procedure
Day 1: Cell Seeding
- Seed HEK293T cells in a 24-well plate at a density of 1 x 10^5 cells per well in complete medium. Incubate overnight to achieve ~70% confluency at transfection.
Day 2: Plasmid Transfection
- For each target site, prepare transfection complexes in an Opti-MEM medium:
- For VPR: Mix 250 ng pLV-dCas9-VPR + 250 ng sgRNA plasmid.
- For SAM: Mix 250 ng pLV-dCas9-VP64 + 125 ng SAM helper plasmid + 125 ng engineered sgRNA plasmid.
- Include controls: "No sgRNA" and "Non-targeting sgRNA."
- Add transfection reagent per manufacturer's instructions. Incubate and add complexes dropwise to cells.
Day 4-5: Harvest and Analysis
- For mRNA-level analysis (qPCR):
- Lyse cells and extract total RNA. Synthesize cDNA.
- Perform qPCR with gene-specific primers. Calculate fold activation (2^(-ΔΔCt)) relative to non-targeting sgRNA control.
- For protein-level analysis (Flow Cytometry, if using reporter):
- Harvest cells, resuspend in flow buffer.
- Analyze GFP fluorescence intensity via flow cytometry. Calculate mean fluorescence intensity (MFI) fold change.
Signaling Pathway and Workflow Diagrams
Diagram 1: CRISPRa Core Recruitment Mechanisms
Diagram 2: CRISPRa Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CRISPRa Research
| Reagent / Material | Function / Description | Example Supplier/Identifier |
|---|---|---|
| dCas9-VPR Expression Plasmid | All-in-one vector expressing the dCas9-VPR fusion protein. | Addgene #63798 (pLV-dCas9-VPR) |
| dCas9-VP64 Expression Plasmid | Core component for the SAM system, expresses dCas9 fused to VP64. | Addgene #61425 (pHRSIN-dCas9-VP64) |
| SAM Helper Plasmid (MS2-P65-HSF1) | Expresses the MCP-p65-HSF1 fusion protein for recruitment via MS2 aptamers. | Addgene #61426 (psPAX2-MS2-P65-HSF1) |
| Engineered sgRNA Cloning Backbone (for SAM) | Vector for expressing sgRNAs with MS2 and PP7 RNA aptamers. | Addgene #73795 (lenti sgRNA-MS2-PP7) |
| Standard sgRNA Cloning Backbone (for VPR) | Vector for expressing a standard, non-aptamer sgRNA. | Addgene #52963 (pU6-sgRNA) |
| Lipofectamine 3000 | High-efficiency transfection reagent for plasmid delivery into mammalian cells. | Thermo Fisher Scientific, L3000015 |
| SYBR Green qPCR Master Mix | For quantitative real-time PCR to measure target gene mRNA levels post-activation. | Applied Biosystems, 4309155 |
| Next-Generation Sequencing Kit | For genomic integrity checks (e.g., GUIDE-seq) and transcriptome analysis (RNA-seq). | Illumina NovaSeq 6000 kits |
| Validated Antibody for Target Protein | For Western Blot analysis to confirm protein-level upregulation. | Cell Signaling Technology, various |
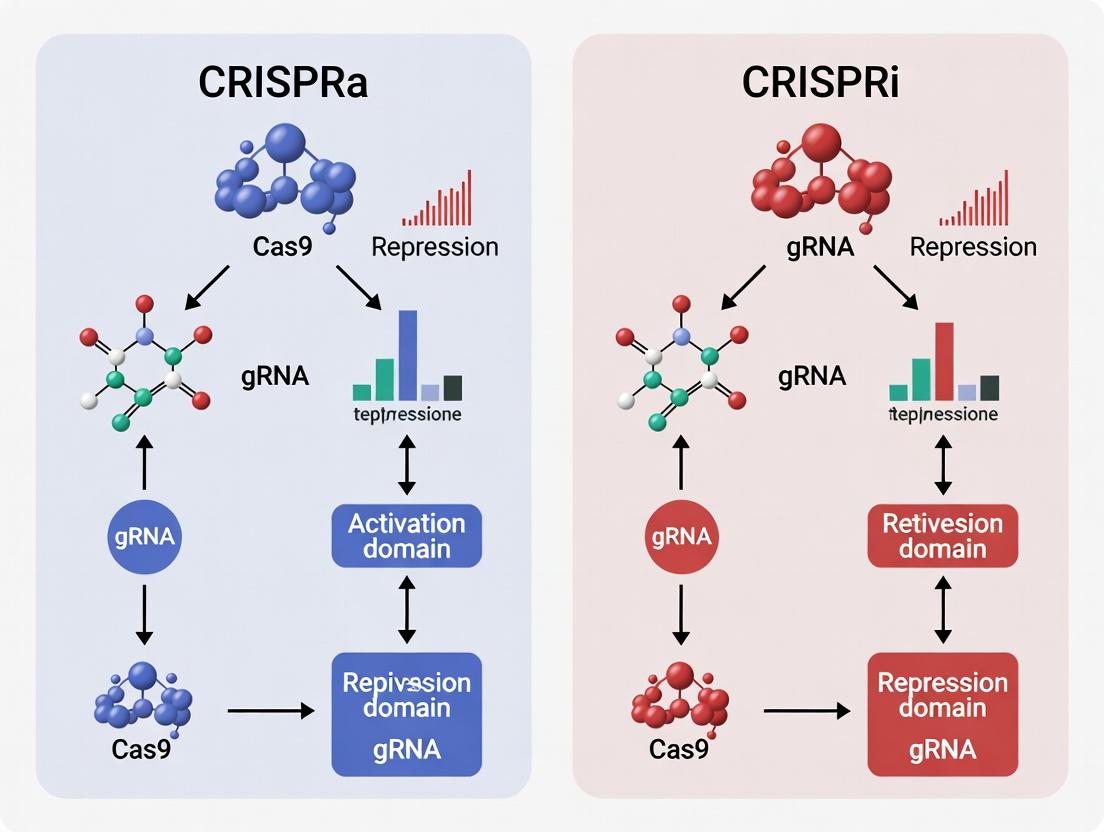
Within the toolkit for programmable gene regulation, CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) represent two foundational pillars. CRISPRi is defined by its precise, targeted transcriptional repression, acting as the functional converse to CRISPRa's gene upregulation. While CRISPRa recruits transcriptional activators to gene promoters, the core mechanism of CRISPRi hinges on the recruitment of dedicated repressive domains to sterically block transcription or compact chromatin. This whitepaper details the molecular architecture and implementation of CRISPRi, focusing on its dominant repressor systems, KRAB and Mxi1, providing a technical guide for their application in functional genomics and therapeutic discovery.
Core Repressor Domains and Their Mechanisms
CRISPRi repressors are typically fused to a catalytically "dead" Cas9 (dCas9), which retains DNA-binding ability but lacks cleavage activity. The choice of repressor domain dictates the downstream silencing mechanism.
KRAB (Krüppel-Associated Box): The most widely used repressor, derived from human zinc-finger proteins. Upon dCas9-KRAB binding to DNA, the KRAB domain recruits endogenous effector proteins, primarily KAP1 (TRIM28). This initiates a cascade of heterochromatin formation:
- KAP1 recruits the SETDB1 histone methyltransferase, which catalyzes H3K9 trimethylation (H3K9me3).
- H3K9me3 recruits HP1 proteins, leading to chromatin compaction and spreading of the repressive state.
- This results in stable, long-term transcriptional silencing.
Mxi1 (Max-interacting protein 1): A mammalian ortholog of the yeast Sin3-interacting domain. dCas9-Mxi1 operates through a distinct, potentially more direct pathway:
- The Mxi1 domain recruits the mammalian Sin3 corepressor complex.
- Sin3 brings histone deacetylases (HDACs), primarily HDAC1 and HDAC2.
- HDACs remove acetyl groups from histone tails (e.g., H3K9ac, H3K27ac), neutralizing transcriptionally active marks and promoting a closed chromatin state.
The quantitative comparison of these systems is summarized in Table 1.
Table 1: Comparison of Core CRISPRi Repressor Systems
| Feature | dCas9-KRAB System | dCas9-Mxi1 System |
|---|---|---|
| Repressor Origin | Human (Zinc finger proteins) | Mammalian (Mad-Max pathway) |
| Primary Effector | KAP1 (TRIM28) | Sin3 corepressor complex |
| Key Enzymatic Activity | Histone Methyltransferase (SETDB1) | Histone Deacetylase (HDAC1/2) |
| Primary Chromatin Mark | H3K9me3 (Repressive) | Loss of H3K9ac/H3K27ac (Active) |
| Silencing Kinetics | Slower, spreading over days | Can be more rapid |
| Typical Repression Fold-Change | 10- to 100-fold (highly dependent on locus) | 5- to 50-fold (highly dependent on locus) |
| Common Applications | Stable gene knock-down, functional screens, epigenetic silencing | Targeted gene silencing, often used in combinatorial setups |
Key Experimental Protocols
Protocol 3.1: Lentiviral Delivery of dCas9-Repressor for Pooled Genetic Screens
This protocol enables genome-wide CRISPRi screening in mammalian cells.
- Library Construction: Clone a genome-wide sgRNA library (e.g., human Brunello library) into a lentiviral vector containing a selection marker (e.g., puromycin resistance).
- Stable Cell Line Generation: a. Produce lentivirus encoding the dCas9-KRAB or dCas9-Mxi1 construct. b. Transduce target cells (e.g., HEK293T, iPSCs) at low MOI (<0.3) and select with appropriate antibiotic (e.g., blasticidin) for 7+ days to generate a polyclonal dCas9-expressing cell line.
- Screen Execution: a. Transduce the dCas9 cell line with the sgRNA library lentivirus at a coverage of >500 cells per sgRNA, maintaining low MOI. b. Select transduced cells (e.g., with puromycin) for 5-7 days. c. Split cells into experimental (e.g., drug treatment) and control arms. Passage cells for 14-21 population doublings.
- Genomic DNA Extraction & Analysis: Harvest cells, extract gDNA, PCR-amplify integrated sgRNA sequences, and quantify by next-generation sequencing. Depletion or enrichment of sgRNAs is analyzed using specialized algorithms (MAGeCK, BAGEL).
Protocol 3.2: Validation of CRISPRi Silencing and Epigenetic State (ChIP-qPCR)
This protocol validates target engagement and chromatin remodeling.
- Cell Preparation: Generate cells expressing dCas9-repressor and a target-specific sgRNA alongside a non-targeting control sgRNA.
- Crosslinking & Sonication: Fix cells with 1% formaldehyde for 10 min. Quench with glycine. Lyse cells and shear chromatin via sonication to ~200-500 bp fragments.
- Immunoprecipitation: Incubate sheared chromatin with antibody-coupled magnetic beads. Key antibodies:
- Validation of dCas9 binding: Anti-Cas9 antibody.
- Validation of KRAB mechanism: Anti-H3K9me3 or anti-KAP1 antibody.
- Validation of Mxi1 mechanism: Anti-H3K9ac or anti-HDAC1 antibody.
- Wash, Elution, and Reverse Crosslink: Wash beads stringently. Elute complexes and reverse crosslinks at 65°C overnight.
- DNA Purification & qPCR: Purify DNA and perform qPCR with primers flanking the sgRNA target site and a control genomic region. Calculate % input or fold enrichment.
Visualizing CRISPRi Mechanisms and Workflows
Title: KRAB Domain Repression Pathway
Title: Mxi1 Domain Repression Pathway
Title: Pooled CRISPRi Screening Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CRISPRi Experiments
| Item | Function & Key Characteristics | Example Vendor/Product |
|---|---|---|
| dCas9-Repressor Plasmids | Mammalian expression vectors for constitutive or inducible expression of dCas9-KRAB or dCas9-Mxi1. | Addgene: #71237 (pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro), #60954 (dCas9-Mxi1) |
| Genome-wide sgRNA Libraries | Pooled, cloned lentiviral libraries targeting all human or mouse genes with multiple sgRNAs per gene. | Broad Institute GPP: Brunello (human), Brie (mouse) libraries. |
| Lentiviral Packaging Plasmids | For safe production of 3rd generation lentivirus (psPAX2, pMD2.G). | Addgene: #12260, #12259 |
| Validated Anti-Histone Antibodies | For ChIP validation of repression marks (H3K9me3) or loss of activation marks (H3K9ac). | Cell Signaling Technology: #13969 (H3K9me3), #9649 (H3K9ac). |
| Next-Generation Sequencing Kit | For high-throughput sequencing of amplified sgRNA regions from genomic DNA. | Illumina: MiSeq Reagent Kit v3. |
| CRISPRi Analysis Software | Bioinformatics tools for identifying essential genes from screen data. | MAGeCK, BAGEL, PinAPL-Py. |
| Inducible Expression Systems | For temporal control of dCas9-repressor or sgRNA expression (e.g., doxycycline-inducible). | Tet-On 3G system, Cumate switch. |
The development of catalytically dead Cas9 (dCas9) has been pivotal in expanding the CRISPR toolkit beyond genome editing into the realms of transcriptional regulation, epigenome engineering, and genomic imaging. This evolution is central to the broader thesis explaining CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) research. While CRISPR-Cas9 for editing relies on Cas9's nuclease activity to create double-strand breaks, dCas9—created through point mutations (e.g., D10A and H840A in S. pyogenes Cas9) that abolish cleavage—retains its ability to bind DNA via gRNA guidance. This creates a programmable, RNA-guided DNA-binding scaffold. In CRISPRi, dCas9 alone sterically blocks transcription initiation or elongation. For CRISPRa, dCas9 serves as a recruitment platform, fused to transcriptional activators (like VP64, p65, Rta) to upregulate gene expression. Thus, dCas9 is the universal foundational component that enables precise, multiplexable, and reversible transcriptional control without altering the DNA sequence.
Core Mechanism and Quantitative Performance
The efficacy of dCas9-based systems is quantified by fold-change in gene expression, specificity, and dynamic range. Key performance metrics for common configurations are summarized below.
Table 1: Performance Metrics of dCas9-Based Transcriptional Modulators
| System | dCas9 Fusion/Recruitment | Typical Fold-Change (Activation/Repression) | Key Applications | Primary Limitations |
|---|---|---|---|---|
| CRISPRi | dCas9 alone (steric block) | Repression: 5- to 100-fold | Gene knockdown, functional genomics | Less effective for genes with high transcription rates. |
| CRISPRi | dCas9-KRAB (repressor domain) | Repression: 10- to 1000-fold | Robust gene silencing, epigenetic silencing | Potential off-target repression. |
| CRISPRa (1st Gen) | dCas9-VP64 | Activation: 2- to 10-fold | Moderate gene upregulation | Limited potency for many mammalian genes. |
| CRISPRa (Synergistic) | dCas9-VPR (VP64-p65-Rta) | Activation: 10- to 1000-fold | Strong gene activation, reprogramming | Increased size may affect delivery. |
| CRISPRa (Recruitment) | SunTag/gRNA scaffold (recruits multiple effectors) | Activation: Up to 2000-fold | Maximal activation, multiplexing | Complex system design and delivery. |
Experimental Protocols for Key dCas9 Applications
Protocol A: CRISPRi for Gene Repression in Mammalian Cells
Objective: To achieve targeted transcriptional knockdown using dCas9-KRAB. Materials: See "The Scientist's Toolkit" below. Procedure:
- Design gRNAs: Design 2-3 gRNAs targeting the promoter or 5' end of the first exon of the gene of interest (typically within -50 to +300 bp relative to TSS). Use established design tools (e.g., CHOPCHOP).
- Clone gRNAs: Clone annealed oligonucleotides encoding the gRNA spacer into your preferred lentiviral gRNA expression vector (e.g., lentiGuide-puro).
- Cell Line Preparation: Generate a stable cell line expressing dCas9-KRAB (e.g., via lentiviral transduction of lenti-dCas9-KRAB-blast and blasticidin selection) or co-transfect/transduce transiently.
- Delivery: Transduce the target cells with the gRNA lentivirus. Include a non-targeting gRNA control.
- Selection & Validation: Apply appropriate antibiotics (e.g., puromycin for gRNA selection) 24h post-transduction. After 72-96 hours, harvest cells for analysis.
- Analysis: Quantify repression via RT-qPCR (for mRNA) and/or western blot (for protein). Normalize to housekeeping genes and non-targeting gRNA control.
Protocol B: CRISPRa for Gene Activation using the SunTag System
Objective: To achieve robust, multiplexed transcriptional activation. Procedure:
- Design gRNAs: Design gRNAs targeting regions -50 to -500 bp upstream of the transcription start site (TSS). Using multiple gRNAs (typically 3-5) per gene enhances efficacy.
- Prepare Cell Line: Establish a stable cell line expressing dCas9 fused to the SunTag peptide array (e.g., dCas9-10xGCN4_v4) and a selectable marker.
- Express scFv-Effector: Co-express a single-chain variable fragment (scFv) antibody fused to a transcriptional activator (e.g., scFv-VP64-p65-Rta) that binds the SunTag. This can be delivered via a separate plasmid or as part of a polycistronic system.
- Deliver gRNAs: Co-transfect the SunTag cell line with plasmids expressing the target-specific gRNAs (cloned into a vector with an appropriate RNA Pol III promoter).
- Analysis: Harvest cells 48-72 hours post-transfection. Assess activation via RT-qPCR, RNA-seq, or functional assays relevant to the target gene.
Visualizing dCas9 Mechanisms in CRISPRa/i
The Scientist's Toolkit: Essential Research Reagents
| Reagent/Material | Function & Explanation |
|---|---|
| dCas9 Expression Vector (e.g., pLV-dCas9-KRAB-blast) | Stably expresses catalytically dead Cas9 fused to an effector domain (like the KRAB repressor). Allows for selection and maintenance of dCas9-expressing cell lines. |
| gRNA Cloning Vector (e.g., lentiGuide-puro) | Backbone for cloning and expressing sequence-specific single guide RNAs (sgRNAs). Contains a selection marker (e.g., puromycin resistance) for enriching transfected/transduced cells. |
| Lentiviral Packaging System (psPAX2, pMD2.G) | Essential for producing lentiviral particles to deliver dCas9 and gRNA constructs into difficult-to-transfect cells, including primary cells. |
| Synergistic Activator Fusion (e.g., VPR: VP64-p65-Rta) | A potent tripartite activator domain fused to dCas9 or a recruitment system to achieve high levels of gene activation. |
| SunTag System Components | A dCas9 fused to a peptide array (SunTag) and a separate scFv-antibody-effector protein (e.g., scFv-VP64). Enables recruitment of multiple activator molecules per dCas9, greatly enhancing potency. |
| Validated Non-Targeting Control gRNA | A gRNA with no perfect match in the host genome. Serves as a critical negative control to account for non-specific effects of dCas9/gRNA expression. |
| RT-qPCR Assay for Target Gene | Validated primers and probes to accurately quantify changes in mRNA expression levels following CRISPRa or CRISPRi perturbation. |
| Next-Generation Sequencing (NGS) Library Prep Kit | For genome-wide assessment of transcriptional outcomes (RNA-seq) or identification of off-target binding sites (e.g., ChIP-seq for dCas9). |
The development of programmable transcriptional regulation, particularly within the CRISPRa (activation) vs. CRISPRi (inhibition) research paradigm, represents a profound evolution from simple, single-component fusion proteins to sophisticated, multi-component systems. This journey mirrors the broader trajectory of synthetic biology and precision therapeutic intervention, moving from proof-of-concept tools to finely tunable platforms capable of complex genetic circuitry.
Historical Progression of CRISPR Regulatory Systems
The foundational principle of fusing a DNA-binding domain to an effector domain was established with zinc finger proteins (ZFPs) and transcription activator-like effectors (TALEs). The advent of CRISPR-Cas9 provided a uniquely programmable and scalable DNA-binding platform, catalyzing rapid innovation.
Phase 1: Early Fusion Proteins (First-Generation CRISPRa/i) The earliest systems involved direct fusion of catalytically dead Cas9 (dCas9) to compact effector domains. For CRISPRi, this was the Krüppel-associated box (KRAB) repressive domain. For CRISPRa, initial attempts used VP64, a tetramer of the herpes simplex viral protein 16. These were simple, one-component systems but offered limited efficacy.
Phase 2: Recruitment of Natural Effector Complexes (Second-Generation) A major leap forward came with systems designed to recruit multiple copies of effectors or endogenous cellular complexes. The SunTag system used an array of peptide epitopes fused to dCas9, which were recognized by single-chain antibody-effector fusions, enabling cooperative recruitment. Similarly, the SAM (Synergistic Activation Mediator) system for CRISPRa used an engineered guide RNA scaffold (MS2 aptamers) to recruit multiple activation proteins (e.g., p65-HSF1), harnessing natural transcriptional machinery.
Phase 3: Advanced Multi-Component & Inducible Systems (Third-Generation) Current state-of-the-art systems incorporate multiple, orthogonal regulatory layers. This includes split-protein systems reconstituted by small molecules, light-inducible dimerization domains (e.g., Cry2/CIB), and logic-gated circuits where multiple guides or Cas proteins are required for activation. These systems allow for precise temporal, spatial, and dose-control over gene expression, critical for therapeutic applications and deciphering complex biological networks.
The core thesis driving this evolution within CRISPRa/i research is the pursuit of specificity, magnitude, and precision in transcriptional control, moving from blunt, constitutive tools to context-aware, dynamic regulators.
Quantitative Evolution of System Performance
Table 1: Performance Metrics Across Generations of CRISPRa Systems
| System Generation | Example System | Typical Fold Activation (Range) | Key Limitation | Primary Innovation |
|---|---|---|---|---|
| Early Fusion (1G) | dCas9-VP64 | 2-10x | Low magnitude, high variability | Proof-of-concept programmability |
| Recruitment Systems (2G) | dCas9-SunTag-VP64, SAM | 10-1000x+ | Larger cargo, potential immunogenicity | Cooperative recruitment, amplified output |
| Advanced Multi-Component (3G) | Light-inducible dCas9-ER/CID, split-dCas9 | Tunable (1-100x+) | Increased complexity | Temporal/spatial control, logic gating |
Table 2: Comparison of Core CRISPRi Repressor Domains
| Repressor Domain | Size (AA approx.) | Mechanism of Action | Typical Repression Efficiency |
|---|---|---|---|
| KRAB (Krüppel-associated box) | ~45 aa | Recruits heterochromatin-forming complexes (e.g., SETDB1, HP1) | 5-50 fold knockdown |
| SID4x (SRF repression domain) | ~100 aa | Recruits co-repressors (e.g., HDACs) via SRF interaction | Comparable to KRAB, context-dependent |
| MeCP2 (methyl-CpG binding domain) | ~85 aa | Binds methylated DNA and recruits repressive complexes | Effective in methylated genomic regions |
Experimental Protocol: Evaluating a Second-Generation CRISPRa System (SAM)
Objective: To assay the transcriptional activation efficiency of the SAM system on a endogenous gene locus in HEK293T cells.
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Guide RNA Design & Cloning: Design three (3) sgRNAs targeting the promoter region ( -50 to -500 bp from TSS) of the gene of interest (GOI). Clone each sgRNA sequence into the MS2-aptamer containing lentiviral sgRNA expression backbone (e.g., lenti-sgRNA(MS2)-zeo).
- Cell Seeding & Transfection: Seed HEK293T cells in a 24-well plate at 1.5 x 10^5 cells/well. After 24 hours, co-transfect using a suitable reagent (e.g., Lipofectamine 3000) with the following plasmids:
- 500 ng of dCas9-VP64 expression plasmid.
- 500 ng of MS2-P65-HSF1 expression plasmid.
- 250 ng of the sgRNA(MS2) plasmid.
- Include controls: a non-targeting sgRNA and a transfection with dCas9-VP64 only.
- Incubation & Harvest: Incubate cells for 48-72 hours to allow for robust gene expression changes.
- RNA Isolation & cDNA Synthesis: Harvest cells and isolate total RNA using a column-based kit. Quantify RNA, and perform reverse transcription with random hexamers to generate cDNA.
- Quantitative PCR (qPCR): Perform qPCR in triplicate using SYBR Green chemistry and primers specific for the GOI and a housekeeping gene (e.g., GAPDH).
- Data Analysis: Calculate fold change in mRNA expression using the ΔΔCt method. Normalize all samples to the housekeeping gene and then to the non-targeting sgRNA control condition.
Key Signaling & Workflow Visualizations
Diagram Title: Evolution of CRISPRa/i System Architectures
Diagram Title: Experimental Protocol for SAM CRISPRa System Evaluation
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CRISPRa/i Functional Studies
| Reagent/Material | Function & Role | Example Product/Catalog |
|---|---|---|
| dCas9-VP64 Expression Plasmid | Core component of 1G/2G CRISPRa; provides programmable DNA binding and basal activation. | Addgene #61425 (pLV dCas9-VP64_Blast) |
| dCas9-KRAB Expression Plasmid | Core component for CRISPRi; provides programmable DNA binding and potent repression. | Addgene #71237 (pHR-dCas9-KRAB-P2A-mCherry) |
| MS2-P65-HSF1 (MCP) Plasmid | Essential second component of SAM system; provides synergistic activation domains. | Addgene #89308 (lenti MS2-P65-HSF1_Hygro) |
| sgRNA(MS2) Cloning Backbone | Vector for expressing sgRNAs with MS2 RNA aptamers to recruit MCP fusion proteins. | Addgene #89307 (lenti sgRNA(MS2)_zeo backbone) |
| Lipofectamine 3000 Transfection Reagent | High-efficiency reagent for plasmid delivery into mammalian cell lines. | Thermo Fisher Scientific L3000015 |
| RNA Isolation Kit | For high-purity total RNA extraction prior to RT-qPCR. | Zymo Research Quick-RNA Miniprep Kit R1055 |
| SYBR Green qPCR Master Mix | Sensitive dye-based chemistry for quantifying mRNA levels. | Bio-Rad iTaq Universal SYBR Green Supermix 1725124 |
| Validated qPCR Primers | Gene-specific primers for target and housekeeping genes. | IDT PrimeTime qPCR Assays (designed per gene) |
From Design to Discovery: Implementing CRISPRa and CRISPRi in Your Research
This technical guide examines the critical choice between CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) for functional genomics studies, forming a core chapter of a broader thesis on "CRISPRa vs CRISPRi explained." The selection of an appropriate perturbation tool is fundamental to experimental validity and impacts subsequent data interpretation in drug discovery and basic research.
Core Mechanisms: A Quantitative Comparison
Table 1: Core Feature Comparison of CRISPRa and CRISPRi Systems
| Feature | CRISPR Activation (CRISPRa) | CRISPR Interference (CRISPRi) |
|---|---|---|
| Primary Function | Transcriptional upregulation (gain-of-function) | Transcriptional repression (loss-of-function) |
| Typical Fold-Change | 2x to >100x induction (varies by locus) | 50% to 90% repression (varies by locus) |
| Catalytic Core | dCas9 (nuclease-dead) fused to activator domains (e.g., VPR, SAM) | dCas9 (nuclease-dead) fused to repressor domains (e.g., KRAB, SID4x) |
| Targeting Specificity | Guide RNA (gRNA) defines genomic locus, typically within ~200 bp upstream of TSS. | Guide RNA (gRNA) defines genomic locus, typically within promoter or early exon. |
| Multiplexing Capacity | High (via pooled gRNA libraries) | High (via pooled gRNA libraries) |
| Common Applications | Genetic suppressor screens, studying gene overexpression phenotypes, synthetic gene circuits. | Essential gene identification, pathway dissection, modeling haploinsufficiency. |
| Key Advantages | Enables study of non-essential genes; can mimic disease-associated overexpression. | Lower off-target effects than RNAi; allows reversible, tunable knockdown. |
| Key Limitations | Magnitude of activation is locus-dependent; potential for artifactual overexpression. | Incomplete knockdown may miss phenotypes; repression efficiency varies. |
Detailed Experimental Protocols
Protocol for a CRISPRa Gain-of-Function Screen (SAM System)
Objective: To perform a positive selection screen for genes whose overexpression confers resistance to a chemotherapeutic agent.
Materials: See Scientist's Toolkit below.
Methodology:
- Library Design & Cloning: Utilize a validated genome-wide CRISPRa gRNA library (e.g., Calabrese et al., 2017). Each gene is targeted by 5-10 gRNAs designed to bind within -200 bp of the transcription start site (TSS). Clone library into the lentiviral SAM vector (containing dCas9-VP64 and MS2-p65-HSF1 activators).
- Virus Production: Generate lentivirus in HEK293T cells by co-transfecting the library plasmid with packaging plasmids psPAX2 and pMD2.G. Harvest supernatant at 48 and 72 hours, concentrate by ultracentrifugation, and titer on target cells.
- Cell Infection & Selection: Infect target cells (e.g., a cancer cell line) at a low MOI (~0.3) to ensure most cells receive a single gRNA. Maintain a representation of >500 cells per gRNA. Select with puromycin for 5-7 days.
- Phenotypic Selection: Split cells into control and treatment groups. Treat with the chemotherapeutic agent at the predetermined IC70 concentration for 14-21 days, refreshing drug and media every 3-4 days.
- Genomic DNA Extraction & NGS: Harvest genomic DNA from pre-selection (T0), control (Tctrl), and treated (Ttreated) populations using a large-scale gDNA kit. Perform PCR amplification of the integrated gRNA cassette using barcoded primers for multiplexing.
- Sequencing & Analysis: Sequence amplicons on an Illumina platform. Align reads to the library reference. Use MAGeCK or similar algorithms to compare gRNA abundance between Ttreated and T0/Tctrl, identifying significantly enriched gRNAs/genes.
Protocol for a CRISPRi Loss-of-Function Screen (dCas9-KRAB)
Objective: To perform a negative selection screen to identify genes essential for cell proliferation.
Materials: See Scientist's Toolkit below.
Methodology:
- Library Design & Cloning: Use a genome-wide CRISPRi library (e.g., Horlbeck et al., 2016) with gRNAs designed to bind from -50 bp of the TSS to the +100 bp of the coding sequence. Clone into a lentiviral vector expressing dCas9-KRAB and the gRNA.
- Virus Production & Cell Infection: As in Protocol 3.1, produce lentivirus and infect target cells at low MOI. Select with appropriate antibiotics.
- Phenotypic Passage: Passage cells continuously for 18-21 population doublings, maintaining representation (>500x coverage). Harvest genomic DNA at the initial time point (T0) and every 3-4 passages (e.g., T7, T14, T21).
- gRNA Amplification & Sequencing: Amplify gRNA sequences from gDNA and prepare for NGS as in Step 5 of Protocol 3.1.
- Analysis: Sequence and align reads. Using MAGeCK, identify gRNAs and genes that drop out significantly in later passages (T21) compared to T0, indicating essentiality.
Visualizing the Systems
Mechanism of CRISPR Activation (SAM System)
Mechanism of CRISPR Interference (dCas9-KRAB)
CRISPRa/i Screening Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPRa and CRISPRi Studies
| Reagent | Function in Experiment | Key Considerations & Examples |
|---|---|---|
| dCas9-Activator Fusion | Core CRISPRa effector. Binds DNA and recruits transcriptional machinery. | VPR: dCas9-VP64-p65-Rta (strong, compact). SAM: dCas9-VP64 with MS2-p65-HSF1 scaffold (very high activation). |
| dCas9-Repressor Fusion | Core CRISPRi effector. Binds DNA and silences transcription. | dCas9-KRAB: Gold standard, recruits histone methyltransferases for stable repression. dCas9-SID4x: Alternative repressor domain. |
| gRNA Expression Vector | Expresses the target-specific guide RNA. | Must be compatible with dCas9 fusion system (e.g., contain MS2 loops for SAM). U6 or H1 promoter driven. |
| Validated gRNA Library | Pooled collection of gRNAs targeting genes genome-wide. | CRISPRa: Designed for regions upstream of TSS. CRISPRi: Designed from -50 to +100 bp relative to TSS. Ensure high coverage (>5 gRNAs/gene). |
| Lentiviral Packaging Plasmids | For production of replication-incompetent lentivirus to deliver constructs. | psPAX2: Provides gag/pol. pMD2.G: Provides VSV-G envelope protein. Third-gen systems enhance safety. |
| Cell Line with dCas9 Stable Expression | A cell line stably expressing the dCas9-activator/repressor. | Enables single-vector delivery of gRNA library only. Critical for consistent screen performance. |
| Next-Generation Sequencing (NGS) Kit | For high-throughput sequencing of gRNA amplicons from genomic DNA. | Must generate sufficient reads to cover library complexity. Illumina platforms are standard. |
| Screen Analysis Software | Computational tool to identify significantly changing gRNAs/genes. | MAGeCK: Robust, accounts for variance. PinAPL-Py: Web-based tool for analysis. |
This technical guide is framed within the broader thesis that CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) are not merely functional opposites but require distinct design paradigms, particularly in guide RNA (gRNA) selection and application. While both systems rely on a catalytically dead Cas9 (dCas9) fused to effector domains, the optimal genomic targeting strategies for transcriptional activation versus repression differ significantly, governed by promoter architecture and the local epigenetic landscape.
Core Principles of gRNA Design for TSS Targeting
Defining the Target Window
The positioning of gRNAs relative to the transcription start site (TSS) is the most critical design parameter. Effective windows are determined by steric constraints of the dCas9-effector complex and the recruitment requirements of the transcriptional machinery.
Table 1: Optimal gRNA Positioning Relative to the TSS
| System | Optimal Target Window (TSS = +1) | Rationale | Key References |
|---|---|---|---|
| CRISPRi (Repression) | -50 to +300 bp (with highest efficacy from +1 to +100) | dCas9 binds DNA and sterically blocks RNA Polymerase II initiation or elongation. Most effective when placed directly within the transcribed region. | (Gilbert et al., Cell 2013); (Qi et al., Cell 2013) |
| CRISPRa (Activation) | -400 to -50 bp (upstream of TSS) | Activation domains (e.g., VPR, SAM) must recruit co-activators to the promoter without interfering with pre-initiation complex assembly. Targeting upstream activator regions is most effective. | (Konermann et al., Nature 2015); (Chavez et al., Nat Methods 2015) |
Epigenetic Context and Chromatin State
The local chromatin environment profoundly impacts dCas9 binding and effector function. Open chromatin (marked by H3K27ac, H3K4me3, DNase I hypersensitivity) facilitates access. Closed, heterochromatic regions (marked by H3K9me3, H3K27me3) hinder binding.
Table 2: Impact of Epigenetic Features on gRNA Efficacy
| Epigenetic Feature | Effect on CRISPRi | Effect on CRISPRa | Design Implication |
|---|---|---|---|
| Open Chromatin (e.g., Active Promoter) | High efficacy; dCas9 binds easily. | High efficacy; activators can engage machinery. | Preferred targeting region for both systems. |
| Repressed/Poised Chromatin (H3K27me3) | Reduced dCas9 binding; partial repression possible. | Very low efficacy; activators cannot overcome Polycomb silencing. | Avoid or pre-treat with chromatin-modifying drugs. |
| Heterochromatin (H3K9me3) | Severely limited dCas9 access. | Negligible activity. | Challenging target; consider alternative epigenome editors. |
| Enhancers (H3K27ac, H3K4me1) | Moderate efficacy (dependent on proximity to TSS). | Can be highly effective for CRISPRa, especially for endogenous enhancer targeting. | Prime targets for activation, especially for gene clusters. |
Diagram 1: Chromatin State Determines gRNA System Feasibility
Detailed Experimental Protocols
Protocol: Identification of Optimal TSS and gRNA Design for CRISPRa/i
Objective: To design and validate high-efficacy gRNAs for a target gene of interest. Materials: See "The Scientist's Toolkit" below. Procedure:
- Define the TSS: Use high-resolution CAGE data or reference databases (e.g., FANTOM5, RefSeq). Do not rely solely on a single annotation; consider alternative TSSs.
- Define the Search Space:
- For CRISPRi: Generate a list of all possible 20-nt guide sequences (NGG PAM) from -50 bp to +300 bp relative to the chosen TSS.
- For CRISPRa: Generate guides targeting -400 bp to -50 bp upstream of the TSS.
- Filter for Specificity: Use algorithms (e.g., CRISPRseek, CHOPCHOP) to score guides for off-target potential. Select guides with ≤3 mismatches in the seed region (positions 1-12) against other genomic sites.
- Score for Efficiency: Predict on-target activity using tools like Rule Set 2 (for SpCas9) or DeepCRISPR. Prioritize guides with high predicted scores.
- Evaluate Epigenetic Context: Overlay candidate guides with public epigenomic datasets (e.g., ENCODE histone modification ChIP-seq, ATAC-seq) for the relevant cell type. Prioritize guides in regions of open chromatin.
- Synthesize and Clone: Clone 3-5 top candidate gRNAs for each gene/target into your delivery vector (lentiviral or all-in-one expression plasmid).
Protocol: Validation of gRNA Efficacy
Objective: To quantitatively measure transcriptional changes induced by CRISPRa/i gRNAs. Procedure:
- Cell Transduction/Transfection: Deliver the dCas9-effector construct (e.g., dCas9-KRAB for i, dCas9-VPR for a) and the individual gRNA constructs into your target cell line. Include a non-targeting control (NTC) gRNA.
- Harvest RNA: 72-96 hours post-delivery, harvest cells and isolate total RNA using a column-based kit with DNase I treatment.
- Quantitative RT-PCR: Perform reverse transcription followed by qPCR using TaqMan or SYBR Green assays specific to the target gene and a housekeeping control (e.g., GAPDH, ACTB).
- Data Analysis: Calculate fold-change using the ΔΔCt method relative to the NTC gRNA condition. For robust conclusions, use ≥3 biological replicates.
- Secondary Validation: For hits, confirm by RNA-seq or via a complementary method (e.g., northern blot, protein-level analysis by western blot).
Diagram 2: gRNA Design and Validation Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPRa/i gRNA Design and Validation
| Reagent/Material | Supplier Examples | Function in Protocol |
|---|---|---|
| dCas9-KRAB Expression Plasmid | Addgene (#71237), Sigma-Aldrich | Core repression effector for CRISPRi experiments. |
| dCas9-VPR or SAM Component Plasmids | Addgene (#63798, #1000000078) | Core activation effector systems for CRISPRa experiments. |
| Lentiviral gRNA Cloning Vector (e.g., lentiGuide-puro) | Addgene (#52963) | For stable integration and expression of gRNAs. |
| High-Fidelity DNA Polymerase (for gRNA cloning) | NEB (Q5), Thermo Fisher | Accurate amplification of gRNA inserts and backbone. |
| Cell Line-Specific Transfection Reagent | Lipofectamine 3000, Fugene HD | Delivery of plasmids to hard-to-transfect cells. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene (#12260, #12259) | For production of lentiviral particles for stable delivery. |
| Polybrene (Hexadimethrine Bromide) | Sigma-Aldrich | Increases lentiviral transduction efficiency. |
| RNA Isolation Kit with DNase I | Qiagen RNeasy, Zymo Research | Prepares pure, genomic DNA-free RNA for qRT-PCR. |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems, Thermo Fisher | Converts RNA to stable cDNA for downstream qPCR. |
| TaqMan Gene Expression Assays | Applied Biosystems, Thermo Fisher | Provides highly specific, probe-based qPCR quantification. |
| Next-Generation Sequencing Service/Library Prep Kit | Illumina, NovaSeq; KAPA HyperPrep | For genome-wide validation via RNA-seq or ChIP-seq. |
The choice of delivery system is a critical determinant for the success of CRISPRa and CRISPRi experiments. Within the broader thesis of comparing transcriptional activation (CRISPRa) and interference (CRISPRi), efficient and sustained delivery of the large dCas9-effector fusion construct (e.g., dCas9-VPR for activation, dCas9-KRAB for repression) is a major technical hurdle. This guide provides a technical comparison of three predominant delivery modalities.
Quantitative Comparison of Delivery Systems
The following table summarizes the core characteristics of each system based on current literature and common practice.
Table 1: Comparative Analysis of Delivery Methods for dCas9-Effectors
| Feature | Lentiviral (LV) | Adeno-Associated Virus (AAV) | Transient Transfection (e.g., PEI, Lipofectamine) |
|---|---|---|---|
| Max Cargo Capacity | ~8-10 kb | ~4.7 kb | Essentially unlimited (multi-plasmid co-transfection) |
| Tropism/Application | Broad, infects dividing & non-dividing cells. | Serotype-dependent; in vivo & in vitro. | Typically in vitro; limited in vivo efficiency. |
| Integration & Duration | Stable genomic integration. Long-term, persistent expression. | Predominantly episomal. Long-term expression in non-dividing cells. | Transient, non-integrating. Expression lasts 48-96 hours. |
| Typical Titer/Throughput | High-titer production (10^8-10^9 IU/mL). Suitable for pooled screens. | High-titer production (10^12-10^13 vg/mL). In vivo applications. | High-throughput, multi-well format. No viral production needed. |
| Immunogenicity | Moderate to high. | Generally low (varies by serotype and host). | Minimal for in vitro chemical methods. |
| Key Advantage | Stable, permanent modification for long-term studies/screens. | Excellent safety profile and in vivo delivery efficiency. | Rapid, flexible, and avoids viral regulatory hurdles. |
| Key Limitation for dCas9-Effectors | Limited capacity for large constructs; risk of insertional mutagenesis. | Severe cargo limit often requires split-intein or dual-vector systems. | Low efficiency in primary/non-dividing cells; transient expression. |
| Best Suited For | Genomic screens, creating stable cell lines for long-term CRISPRa/i. | In vivo gene regulation studies, clinical applications. | Rapid proof-of-concept, easily multiplexed experiments in amenable cell lines. |
Detailed Methodologies and Protocols
Lentiviral Production and Transduction for dCas9-Effector Delivery
This protocol is for creating a stable cell line expressing a dCas9-effector.
Materials:
- Packaging plasmids (psPAX2, pMD2.G)
- Transfer plasmid encoding dCas9-VPR or dCas9-KRAB and a selection marker (e.g., puromycin resistance)
- HEK293T cells (for virus production)
- Target cells (e.g., HeLa, primary fibroblasts)
- Polyethylenimine (PEI) Max transfection reagent
- Serum-containing media for 293T and target cells
- 0.45 µm PVDF filter
- Polybrene (hexadimethrine bromide, 8 µg/mL final concentration)
- Appropriate selection antibiotic (e.g., puromycin)
Procedure:
- Day 1: Seed HEK293T cells in a 10 cm dish to reach 70-80% confluency the next day.
- Day 2: Transfect cells using PEI Max. Prepare two tubes: Tube A with 10 µg transfer plasmid, 7.5 µg psPAX2, and 2.5 µg pMD2.G in 500 µL serum-free media. Tube B with 45 µL PEI Max in 500 µL serum-free media. Combine after 5 min, incubate 20 min, and add dropwise to cells.
- Day 3: Replace media with fresh complete media.
- Day 4 & 5: Harvest viral supernatant (~48 and 72 hours post-transfection), filter through a 0.45 µm filter, and either use immediately or aliquot and store at -80°C.
- Transduction: Seed target cells in a 6-well plate. The next day, add viral supernatant and Polybrene (8 µg/mL). Spinoculate by centrifuging at 800-1000 x g for 30-60 min at 32°C, then return to incubator.
- Day 6: Replace media with fresh complete media.
- Day 7: Begin selection with the appropriate antibiotic (e.g., 1-2 µg/mL puromycin). Change media with antibiotic every 2-3 days until all non-transduced control cells are dead (typically 3-7 days).
AAV Production via PEI Transfection (Serotype 2/8/9)
Protocol for producing AAV vectors, often requiring a dual-vector system for large dCas9-effectors.
Materials:
- AAV transfer plasmid (containing ITRs, promoter, and gene of interest—e.g., split-dCas9-effector)
- AAV helper plasmid (pHelper)
- AAV Rep/Cap plasmid (e.g., serotype 2, 8, or 9)
- HEK293T cells
- PEI Max transfection reagent
- Dulbecco's Modified Eagle Medium (DMEM) with serum
- 150 mM NaCl solution
- Benzonase nuclease
- Ammonium sulfate
- Iodixanol gradient solutions (15%, 25%, 40%, 60% in PBS-MK)
- Ultracentrifuge and tubes
Procedure:
- Day 1: Seed HEK293T cells in fifteen 15 cm dishes.
- Day 2: Transfect per dish with a 1:1:1 molar ratio of AAV transfer plasmid, Rep/Cap plasmid, and pHelper plasmid using PEI Max. Total DNA per dish is typically 20 µg.
- Day 5 (72h post-transfection): Harvest cells by scraping and pellet by centrifugation. Resuspend pellet in 150 mM NaCl, freeze-thaw three times, and treat with Benzonase (50 U/mL) for 30 min at 37°C.
- Clarify the lysate by centrifugation and precipitate the supernatant with equal volume of 40% PEG8000/1M NaCl. Incubate on ice for 1h.
- Pellet the PEG precipitate, resuspend in PBS, and layer onto a pre-formed iodixanol step gradient (15%, 25%, 40%, 60%) in a Beckman quick-seal tube.
- Ultracentrifuge at 350,000 x g for 1-2 hours at 18°C.
- Collect the opaque 40% iodixanol fraction containing purified AAV particles. Dialyze against PBS + 5% glycerol, concentrate, and titer via qPCR.
Transient Transfection of dCas9-Effector and sgRNA Plasmids
A standard protocol for rapid, transient expression in HEK293T cells.
Materials:
- dCas9-effector plasmid (e.g., pLV-dCas9-VPR)
- sgRNA expression plasmid (e.g., pU6-sgRNA-EF1a-Puro)
- Adherent cells (e.g., HEK293T, HeLa)
- Lipofectamine 3000 reagent
- Opti-MEM Reduced Serum Media
- Appropriate media for cell line
Procedure:
- Day 1: Seed cells in a 24-well plate to reach 70-90% confluency at transfection.
- Day 2: For each well, prepare two tubes:
- Tube A (DNA Mix): Dilute 500 ng total DNA (e.g., 250 ng dCas9-effector + 250 ng sgRNA plasmid) in 25 µL Opti-MEM. Add 1 µL P3000 Enhancer Reagent.
- Tube B (Lipid Mix): Dilute 1.5 µL Lipofectamine 3000 in 25 µL Opti-MEM. Incubate 5 min.
- Combine Tube A and B, mix gently, incubate for 15-20 min at RT.
- Add the 50 µL DNA-lipid complex dropwise to the well containing 500 µL complete media.
- Day 3 (24h post-transfection): Replace media with fresh complete media.
- Day 4-5: Assay for CRISPRa/i effects (e.g., RT-qPCR, RNA-seq). Expression peaks ~48-72 hours post-transfection.
Visualizations
Title: Decision Workflow for dCas9-Effector Delivery System
Title: Cargo Capacity and Configurations for Each Delivery System
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for dCas9-Effector Delivery Experiments
| Reagent/Material | Primary Function | Key Consideration |
|---|---|---|
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Provide gag/pol and VSV-G envelope proteins in trans for virus production. | Second/third generation systems improve safety. |
| AAV Rep/Cap Plasmid | Provides AAV replication (Rep) and capsid (Cap) proteins for viral packaging. | Serotype (e.g., 2, 8, 9, DJ) determines tropism and efficiency. |
| Polyethylenimine (PEI) Max | Cationic polymer for transient transfection of viral packaging or plasmid DNA. | Cost-effective at scale for viral production. |
| Lipofectamine 3000 | Lipid-based transfection reagent for transient delivery of plasmids. | High efficiency in many immortalized cell lines. |
| Polybrene | Cationic polymer that neutralizes charge repulsion between virus and cell membrane. | Increases transduction efficiency for lentivirus. |
| Puromycin Dihydrochloride | Aminonucleoside antibiotic for selecting cells stably expressing resistance genes. | Kill curve must be established for each cell line. |
| Iodixanol | Density gradient medium for purifying AAV particles away from cellular debris. | Non-ionic, iso-osmotic, and preserves virus infectivity. |
| Benzonase Nuclease | Degrades unpackaged viral genomes and contaminating cellular nucleic acids. | Crucial for reducing viscosity and improving AAV purity. |
| qPCR Kit for Viral Titering | Quantifies viral genome copies (vg/mL) for AAV or lentiviral vector copies. | Requires a standard curve from a plasmid of known concentration. |
This whitepaper details the implementation of genome-wide CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) screens, positioned within the broader analytical thesis of CRISPRa vs CRISPRi explained research. The core distinction lies in their mode of transcriptional regulation: CRISPRa uses a modified, catalytically dead Cas9 (dCas9) fused to transcriptional activators (e.g., VPR, SAM) to upregulate gene expression, while CRISPRi employs dCas9 fused to transcriptional repressors (e.g., KRAB) to downregulate expression. The choice between them is determined by the biological question—whether gain-of-function or loss-of-function phenotypes are sought—and is influenced by factors like efficiency, specificity, and technical requirements.
Core System Architectures and Quantitative Comparisons
Key System Components
- CRISPRi: dCas9-KRAB (Krüppel-associated box) fusion. KRAB recruits endogenous repressive complexes (e.g., SETDB1, HP1) to promote heterochromatin formation.
- CRISPRa: Multiple architectures exist, with varying potency:
- VP64: dCas9 fused to a tetramer of the VP16 activation domain. Moderate activity.
- VPR: dCas9 fused to a tripartite activator (VP64-p65-Rta). High activity.
- SAM (Synergistic Activation Mediator): A three-component system involving dCas9-VP64, an engineered MS2-p65-HSF1 activator recruited via sgRNA stem-loops (MS2), and the co-expressed MCP protein. Very high activity.
Quantitative Performance Data
Table 1: Comparative Performance of CRISPRa and CRISPRi Systems
| Parameter | CRISPRi (dCas9-KRAB) | CRISPRa (VPR) | CRISPRa (SAM) |
|---|---|---|---|
| Typical Repression/Activation | 70-95% knockdown | 5-50x activation | 100-1000x activation |
| Optimal Targeting Region | -50 to +300 bp relative to TSS | -50 to -500 bp upstream of TSS | -50 to -500 bp upstream of TSS |
| Library Size (Genome-wide) | ~4-5 sgRNAs per gene (~90,000 total) | ~4-10 sgRNAs per gene (~100,000 total) | ~4-10 sgRNAs per gene (~100,000 total) |
| Background Noise | Low | Moderate | Higher (potential for off-target activation) |
| Key Advantage | High knockdown consistency, low background | Simpler single-vector system | Maximum activation potency |
Experimental Protocol for a Pooled Genome-wide Screen
A typical pooled screen involves transducing a population of cells with a lentiviral sgRNA library at low MOI, selecting for stably integrated cells, applying a selective pressure (e.g., drug, FACS), and sequencing the sgRNA barcodes to determine enrichment/depletion.
Detailed Step-by-Step Methodology
Part A: Library Preparation and Lentivirus Production
- Acquire Library: Obtain a commercially available genome-wide CRISPRa or CRISPRi lentiviral sgRNA library (e.g., Calabrese, SAM, or CRISPRi v2 libraries from Addgene).
- Amplify Library: Transform the plasmid library into Endura electrocompetent E. coli. Plate on large 24 x 24 cm bioassay dishes with carbenicillin. Harvest colonies via scraping and perform Maxiprep plasmid DNA isolation. Critical: Maintain >500x library representation at each amplification step.
- Produce Lentivirus: Co-transfect HEK293T cells (in 10-layer cell factories) with the sgRNA library plasmid, psPAX2 (packaging), and pMD2.G (VSV-G envelope) plasmids using PEI transfection reagent.
- Harvest and Titrate: Collect viral supernatant at 48 and 72 hours post-transfection, concentrate via ultracentrifugation, and titrate on target cells using puromycin selection or qPCR.
Part B: Cell Line Engineering and Screening
- Generate Stable Cells: Stably express dCas9-activator or dCas9-repressor in your target cell line using lentivirus and blasticidin selection. Validate expression by western blot and functional assay.
- Library Transduction: Transduce the engineered cells with the sgRNA library virus at an MOI of ~0.3-0.4 to ensure >95% of cells receive a single sgRNA. Use a minimum of 500 cells per sgRNA in the population (e.g., for a 100k library, transduce 50 million cells).
- Puromycin Selection: 24-48 hours post-transduction, add puromycin (for libraries with puromycin resistance) for 5-7 days to select transduced cells.
Part C: Screening and Analysis
- Apply Selection/Assay Phenotype: Split cells into control and experimental arms. Apply the selective pressure (e.g., toxin, nutrient stress, infectivity) or use FACS to isolate populations based on a reporter (e.g., GFP expression).
- Harvest Genomic DNA: At the experimental endpoint (typically 14-21 days post-selection), harvest cell pellets (maintaining representation). Extract genomic DNA using a Maxiprep scale kit.
- Amplify sgRNA Barcodes: Perform a two-step PCR to amplify the sgRNA cassette from genomic DNA and add Illumina sequencing adapters/indexes.
- Next-Generation Sequencing (NGS): Pool PCR products and sequence on an Illumina NextSeq or HiSeq platform (75-100 bp single-end reads).
- Bioinformatic Analysis: Align reads to the sgRNA library reference. Use MAGeCK or PinAPL-Py algorithms to statistically compare sgRNA abundance between control and experimental samples, identifying significantly enriched or depleted sgRNAs and their target genes.
Visualization of Experimental Workflow and Mechanism
Diagram 1: Pooled CRISPRa/i Screen Workflow
Diagram 2: CRISPRi vs. CRISPRa (SAM) Molecular Mechanism
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for CRISPRa/i Screens
| Reagent/Material | Supplier Examples | Function & Critical Notes |
|---|---|---|
| Genome-wide sgRNA Library (CRISPRa or CRISPRi) | Addgene (Human SAM, Calabrese), Sigma (MISSION), Dharmacon | Pre-designed, cloned lentiviral pools targeting all human or mouse genes. Ensure compatibility with your dCas9 system. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene | Second and third-generation packaging plasmids required for producing replication-incompetent lentivirus. |
| dCas9-Activator/Repressor Plasmid | Addgene (lentidCas9-VPR, lenti dCas9-KRAB-blast) | Stable expression vector for the core transcriptional regulator. Contains selection marker (e.g., blasticidin). |
| Lentiviral Transfection Reagent (PEI or Lipid-based) | Polysciences (PEI MAX), Thermo Fisher (Lipofectamine 3000) | For high-efficiency co-transfection of library and packaging plasmids into HEK293T cells. |
| HEK293T/HEK293FT Cell Line | ATCC | Standard cell line for high-titer lentivirus production due to high transfection efficiency and permissiveness. |
| Puromycin, Blasticidin S HCl | Thermo Fisher, Invivogen | Antibiotics for selection of transduced cells (puromycin) or cells stably expressing dCas9 (blasticidin). Titrate for each cell line. |
| Next-Gen Sequencing Kit (MiSeq/NextSeq) | Illumina | For high-throughput sequencing of sgRNA amplicons. 75-cycle kits are standard. |
| gDNA Extraction Kit (Maxi/Midi Prep) | Qiagen (Blood & Cell Culture DNA Maxi), Promega | For large-scale, high-quality genomic DNA extraction from millions of screened cells. |
| sgRNA Amplification PCR Primers & Master Mix | IDT, NEB | Custom primers with Illumina adapters and high-fidelity polymerase for specific, unbiased amplification of sgRNA regions from gDNA. |
| Bioinformatics Software (MAGeCK) | Open Source (https://sourceforge.net/p/mageck) | Essential computational tool for analyzing screen data, normalizing counts, and identifying significantly enriched/depleted genes using robust statistical models (RRA algorithm). |
The advent of CRISPR-based transcriptional modulation—specifically CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi)—has revolutionized therapeutic and translational research. While both systems utilize a catalytically dead Cas9 (dCas9) to target specific genomic loci, their functional outputs are diametrically opposed. CRISPRa recruits transcriptional activators to enhance gene expression, whereas CRISPRi recruits repressors to silence it. This precise control over gene expression levels provides a powerful toolkit for modeling polygenic diseases, performing functional genomics for drug target discovery, and directing cellular reprogramming for regenerative medicine. This whitepaper details the technical application of these systems within these three critical translational domains.
Core Quantitative Comparison: CRISPRa vs. CRISPRi
The table below summarizes the key quantitative parameters differentiating CRISPRa and CRISPRi systems, based on recent pooled screen data (2023-2024).
Table 1: Performance Metrics of CRISPRa vs. CRISPRi in Human Cells
| Parameter | CRISPRa (e.g., dCas9-VPR) | CRISPRi (e.g., dCas9-KRAB) |
|---|---|---|
| Typical Gene Induction | 5x - 1,000x (context-dependent) | Not Applicable |
| Typical Gene Repression | Not Applicable | 70% - 95% knockdown |
| Optimal Targeting Region | -200 to -50 bp from TSS | -50 to +300 bp from TSS |
| Multiplexing Capacity | High (with arrayed gRNAs) | High (with arrayed gRNAs) |
| Off-Target Effects | Moderate (epigenetic seeding) | Moderate (epigenetic seeding) |
| Screen Hit Rate (Gain-of-Function) | 2-5% of library | Not Primary Application |
| Screen Hit Rate (Loss-of-Function) | Not Primary Application | 1-3% of library |
| Primary Application in Screens | Resistance mechanisms, enhancer mapping | Essential gene identification, vulnerability discovery |
Modeling Complex Diseases
CRISPRa/i enables the establishment of more accurate in vitro and in vivo disease models by modulating the expression of disease-associated genes or risk alleles without altering the DNA sequence.
Experimental Protocol: Creating a Polygenic Disease Model Using CRISPRa
Objective: To model a polygenic neurodegenerative disease (e.g., Parkinson's) by simultaneously overexpressing three risk genes (SNCA, LRRK2, GBA1) in human induced pluripotent stem cell (iPSC)-derived neurons.
- Design gRNAs: Design three sgRNAs per target gene, targeting positions -150 bp upstream of the transcriptional start site (TSS). Use validated algorithms (e.g., CRISPick).
- Construct Lentiviral Vectors: Clone each sgRNA into a lentiviral plasmid containing a dCas9-VPR (for CRISPRa) expression cassette. Include unique barcodes for each sgRNA.
- Produce Virus: Generate high-titer lentivirus for each sgRNA pool in HEK293T cells.
- Transduce Cells: Transduce iPSC-derived dopaminergic neuronal progenitors with a pooled lentiviral mix at a low MOI (<0.3) to ensure single integration.
- Select and Culture: Select transduced cells with puromycin (2 μg/mL) for 7 days. Differentiate progenitors into mature neurons over 4-6 weeks.
- Validate Model: Quantify gene expression via qRT-PCR and protein via Western blot. Assess disease phenotypes: α-synuclein aggregation (immunofluorescence), neuronal activity (MEA), and survival (CellTiter-Glo).
Visualization: Workflow for Polygenic Disease Modeling
Drug Target Discovery via Functional Genomics
Genome-wide CRISPRa and CRISPRi screens are indispensable for identifying and validating novel drug targets.
Experimental Protocol: Genome-wide CRISPRi Resistance Screen
Objective: Identify genes whose knockdown confers resistance to a chemotherapeutic agent (e.g., Doxorubicin) in a breast cancer cell line (MCF-7).
- Library Transduction: Transduce MCF-7 cells stably expressing dCas9-KRAB with a genome-wide CRISPRi lentiviral sgRNA library (e.g., hCRISPRi-v2) at an MOI of ~0.3 to ensure single guide integration. Maintain >500x library representation.
- Selection and Split: After puromycin selection, split cells into two arms: Treatment Arm (200 nM Doxorubicin) and Control Arm (DMSO).
- Passaging: Culture cells for 14-16 population doublings, maintaining drug pressure and library coverage.
- Genomic DNA Extraction & Sequencing: Harvest genomic DNA from both arms at endpoint. Amplify integrated sgRNA sequences via PCR and subject to high-throughput sequencing (Illumina).
- Bioinformatic Analysis: Align sequences to the reference library. Use MAGeCK or similar tools to compare sgRNA abundance between treatment and control arms. Genes enriched with multiple sgRNAs in the treatment arm are candidate resistance targets.
Table 2: Key Reagents for CRISPRi Resistance Screen
| Reagent / Material | Function & Explanation |
|---|---|
| dCas9-KRAB Stable Cell Line | Provides consistent, inducible transcriptional repression machinery. |
| Genome-wide CRISPRi Library | Pooled lentiviral sgRNAs targeting all human genes (5-10 sgRNAs/gene). |
| Lentiviral Packaging Mix | Third-generation system (psPAX2, pMD2.G) for safe, high-titer virus production. |
| Polybrene (8 μg/mL) | Enhances viral transduction efficiency in mammalian cells. |
| Puromycin Dihydrochloride | Selects for cells successfully transduced with the sgRNA library. |
| Doxorubicin Hydrochloride | The chemotherapeutic agent providing selective pressure in the screen. |
| CellTiter-Glo 3D Assay | Quantifies cell viability/cytotoxicity in a luminescent format. |
| Next-Generation Sequencing Kit | For preparing and sequencing the amplified sgRNA barcodes. |
Visualization: Key Signaling Pathways in Identified Targets
Cellular Reprogramming and Transdifferentiation
CRISPRa/i enables direct lineage reprogramming by modulating master regulator genes, bypassing pluripotent states.
Experimental Protocol: Direct Cardiac Reprogramming Using CRISPRa
Objective: Transdifferentiate human dermal fibroblasts into induced cardiomyocyte-like cells (iCMs) by overexpressing cardiac factors (GATA4, MEF2C, TBX5).
- Design and Clone: Design sgRNAs targeting promoters of GATA4, MEF2C, and TBX5. Clone them into a lentiviral vector with dCas9-VPR and a fluorescent reporter.
- Transduce Fibroblasts: Plate primary human fibroblasts and transduce with the pooled CRISPRa virus in the presence of polybrene.
- Media Switch: 48h post-transduction, switch to cardiac induction media (supplemented with small molecules like CHIR99021 and Ascorbic Acid).
- Monitor and Enrich: Monitor for reporter expression and morphological changes. After 14 days, enrich for iCMs via glucose deprivation media or fluorescence-activated cell sorting (FACS).
- Characterization: Assess functional markers: cardiac Troponin T (cTnT) immunofluorescence, spontaneous contraction (video microscopy), and cardiac gene expression panel (RNA-seq).
Visualization: Logical Framework for Direct Reprogramming
Maximizing Efficacy: Troubleshooting Common Pitfalls in CRISPRa/i Experiments
Within the broader thesis comparing CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), a critical operational challenge is suboptimal phenotypic outcomes—specifically, low target gene activation or incomplete repression. This guide posits that the two most determinative, and often interrelated, factors governing efficacy are guide RNA (gRNA) design efficiency and local chromatin accessibility. While CRISPRa and CRISPRi utilize distinct effector domains (e.g., VP64-p65-Rta for activation; KRAB, SID4X for repression) to modulate transcription, both systems are fundamentally constrained by the ability of the catalytically dead Cas9 (dCas9)-effector complex to physically occupy its target DNA site. This occupancy is a prerequisite for function and is highly sensitive to gRNA:DNA hybridization kinetics and the nucleosome occupancy of the target locus.
Core Determinants of Efficacy
Guide RNA (gRNA) Efficiency
gRNA efficiency is governed by sequence-specific features beyond simple complementarity to the target. Key determinants include:
- On-Target Scoring: GC content (optimal 40-60%), avoidance of homopolymers, and position-specific nucleotide preferences, particularly at the PAM-distal seed region (nucleotides 1-10).
- Off-Target Potential: Mismatch tolerance, especially in the seed region, which can lead to dCas9 binding at non-target loci, diluting the effective pool of effector complexes.
- Secondary Structure: Self-complementarity within the gRNA spacer or scaffold that can inhibit Cas9 binding.
Table 1: Key Features for Predictive gRNA Efficiency Scoring
| Feature | Optimal Range/Characteristic | Impact on CRISPRa/i |
|---|---|---|
| GC Content | 40% - 60% | High GC increases stability but may reduce specificity; low GC reduces binding affinity. |
| Seed Region (pos 1-10) | High specificity, no mismatches | Critical for initial DNA recognition; mismatches here drastically reduce binding. |
| Terminal Nucleotide (pos 20) | Avoid 'G' at 5' end (T7 promoter) | Can interfere with transcription initiation for U6-expressed gRNAs. |
| Off-Target Mismatches | >3 mismatches, especially in seed | Determines specificity; influences signal-to-noise ratio in modulation. |
| Predicted On-Target Score | >60 (tool-dependent) | Aggregate metric from algorithms like Rule Set 2, CRISPRon, or DeepHF. |
Chromatin Accessibility
The eukaryotic genome is packaged into chromatin, with nucleosomes acting as primary barriers to dCas9 binding. Open Chromatin Regions (OCRs), often marked by DNase I hypersensitivity or H3K27ac, are permissive. Closed chromatin, marked by H3K9me3 or H3K27me3, is restrictive.
- CRISPRa Dependence: CRISPRa is exceptionally sensitive to chromatin state. Activators must not only bind but also recruit chromatin remodelers to initiate transcription. A closed target site often leads to complete failure.
- CRISPRi Robustness: KRAB-based repressors can spread heterochromatin, making repression potentially more effective even at moderately accessible sites, but initial binding remains a barrier.
Table 2: Chromatin Features Affecting dCas9-Effector Binding
| Chromatin Feature | Assay/Modification | Implication for CRISPRa/i |
|---|---|---|
| Open Chromatin | ATAC-seq, DNase-seq, H3K27ac | Permissive: High probability of dCas9 binding. Essential for CRISPRa initiation. |
| Promoter State | H3K4me3 (active), H3K27me3 (poised) | Active promoters (H3K4me3) are more responsive to CRISPRa/i than poised or inactive. |
| Heterochromatin | H3K9me3, H3K27me3 | Restrictive: Physically blocks dCas9 binding. Major cause of failure. |
| Nucleosome Position | MNase-seq, NucleoATAC | Target sites within nucleosome cores are occluded; linker regions are accessible. |
Diagram Title: Determinants of dCas9-Effector Binding and Function
Diagnostic Experimental Workflow
A systematic approach is required to diagnose the cause of low activity.
Workflow 1: Diagnostic Pipeline for Low CRISPRa/i Efficiency
Diagram Title: Diagnostic Workflow for Low CRISPRa/i Efficiency
Detailed Protocols
Protocol A: Validating dCas9 Occupancy via ChIP-qPCR Objective: Quantify the amount of dCas9 bound to the target site relative to a control site.
- Crosslinking & Lysis: Harvest 5x10^6 cells transfected with dCas9-effector + gRNA. Crosslink with 1% formaldehyde for 10 min at RT. Quench with 125mM glycine. Lyse cells in ChIP lysis buffer.
- Chromatin Shearing: Sonicate lysate to shear DNA to 200-500 bp fragments. Confirm fragment size by agarose gel electrophoresis.
- Immunoprecipitation: Incubate chromatin with 2-5 µg of anti-FLAG (for tagged dCas9) or anti-Cas9 antibody overnight at 4°C. Use Protein A/G magnetic beads for capture. Include an IgG isotype control.
- Washes & Elution: Wash beads sequentially with Low Salt, High Salt, LiCl, and TE buffers. Elute chromatin with elution buffer (1% SDS, 100mM NaHCO3) and reverse crosslinks at 65°C overnight.
- qPCR Analysis: Purify DNA and perform qPCR using primers flanking the gRNA target site and a control region (e.g., GAPDH). Calculate % input and fold enrichment over control gRNA sample.
Protocol B: Assessing Local Chromatin Accessibility via ATAC-seq (Lite Protocol) Objective: Map open chromatin regions in your cell population.
- Nuclei Preparation: Wash 50,000 viable cells in cold PBS. Lyse in ATAC-seq lysis buffer (10mM Tris-Cl pH7.4, 10mM NaCl, 3mM MgCl2, 0.1% IGEPAL CA-630). Pellet nuclei immediately.
- Tagmentation: Resuspend nuclei in transposase reaction mix (Illumina Nextera Tn5). Incubate at 37°C for 30 min. Immediately purify DNA using a MinElute PCR Purification Kit.
- Library Amplification: Amplify tagmented DNA with 10-12 cycles of PCR using indexed primers. Clean up library with SPRI beads.
- Data Analysis: After sequencing, align reads to the reference genome. Call peaks (accessible regions) using MACS2. Visualize signal at your target locus in IGV. Compare to public DNase/ATAC datasets (e.g., ENCODE).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Diagnosing CRISPRa/i Efficiency
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Validated dCas9-Effector Plasmid | Expresses the core fusion protein (e.g., dCas9-VPR, dCas9-KRAB). Use a well-validated backbone (e.g., Addgene #) for consistency. | Addgene #xxxxx (dCas9-VPR), #yyyyy (dCas9-KRAB) |
| gRNA Cloning Vector | Backbone for expressing gRNA under U6 promoter. Allows rapid cloning of spacer sequences. | Addgene #41824 (px459), #47108 (gRNA_Cloning Vector) |
| ChIP-Grade Antibody | High-specificity antibody for immunoprecipitating dCas9 (e.g., anti-FLAG M2, anti-HA, anti-Cas9). Critical for occupancy assays. | Sigma F1804 (Anti-FLAG M2), Cell Signaling #14697 (Anti-Cas9) |
| ATAC-seq Kit | Optimized reagents for nuclei preparation and tagmentation. Ensures reproducible open chromatin profiling. | Illumina (Nextera DNA Library Prep), 10x Genomics (Chromium Next GEM) |
| Chromatin Accessibility Data | Public or commercial reference datasets (ATAC/DNase-seq) for your cell type. Essential for pre-screening target sites. | ENCODE Portal, Cistrome DB, UCSC Genome Browser |
| gRNA Design & Scoring Tool | Algorithm to predict on-target efficiency and off-targets. Informs initial design and diagnosis. | Broad Institute GPP Portal (CRISPRscan), ChopChop, CRISPick |
| Positive Control gRNA | gRNA targeting a highly accessible, constitutively expressed locus (e.g., CCR5, AAVS1). Serves as a transfection and binding control. | Synthesized oligos targeting AAVS1 safe harbor. |
| qPCR Primers for Target Locus | Validated primers for ChIP-qPCR or RT-qPCR. Must flank the gRNA cut site but be outside the edited region. | Custom-designed, NCBI Primer-BLAST validated. |
Within the evolving landscape of CRISPR-based functional genomics, the distinction between CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) is fundamental. CRISPRa aims to upregulate gene expression, typically by recruiting transcriptional activators like VPR or SAM complex to a promoter region. Conversely, CRISPRi represses transcription, often via a catalytically dead Cas9 (dCas9) fused to repressive domains such as KRAB. While both are precise alternatives to genome editing, their reliance on programmable DNA binding introduces the persistent risk of off-target effects, where the guide RNA (gRNA) directs the complex to genomic loci with incomplete complementarity. This can lead to erroneous gene modulation, confounding experimental results and posing significant safety risks in therapeutic contexts. This whitepaper provides an in-depth technical guide to understanding, measuring, and mitigating these specificity concerns.
Quantitative Landscape of Off-Target Effects in CRISPRa/i
Recent studies have benchmarked the specificity of various CRISPRa and CRISPRi systems. Key quantitative findings are summarized below.
Table 1: Comparative Off-Target Profiling of Major Transcriptional Modulators
| System (dCas9 Fusion) | Primary Function | Typical On-Target Fold-Change | Median Off-Target Fold-Change | Key Off-Target Assessment Method | Reference (Year) |
|---|---|---|---|---|---|
| dCas9-KRAB | CRISPRi (Repression) | 5-10x downregulation | 1.5-2x downregulation | ChIP-seq & RNA-seq | (Gilbert et al., 2014) |
| dCas9-VPR | CRISPRa (Activation) | 50-100x upregulation | 2-5x upregulation | GUIDE-seq & RNA-seq | (Chavez et al., 2016) |
| dCas9-SAM | CRISPRa (Activation) | 100-500x upregulation | 3-8x upregulation | BLISS & scRNA-seq | (Konermann et al., 2018) |
| dCas9-p300 Core | Epigenetic Activation | 20-50x upregulation | 1.5-3x upregulation | CIRCLE-seq & ChIP-seq | (Hilton et al., 2015) |
| CRISPRoff/on (ZNF fused) | Epigenetic Silence/Activation | Stable >10x repression/activation | <2x (highly specific) | Whole-genome bisulfite sequencing | (Núñez et al., 2021) |
Table 2: Factors Influencing Specificity & Their Quantitative Impact
| Factor | High-Specificity Condition | Low-Specificity Condition | Effect on Off-Target Rate (Relative Increase) |
|---|---|---|---|
| gRNA Length | 20-nt + 5' G (for SpCas9) | 17-nt truncated | 3-5x |
| GC Content | 40-60% | <20% or >80% | 2-4x |
| gRNA Delivery | Lentiviral (low copy) | Transient (high plasmid) | 2-3x |
| dCas9 Expression | Low, Inducible | Constitutive, High | 4-8x |
| Chromatin State | Open (DNase I hypersensitive) | Closed (Heterochromatin) | Variable (On-target efficiency drops) |
Core Experimental Protocols for Assessing Specificity
Protocol 3.1: Genome-Wide Off-Target Detection Using GUIDE-seq
Objective: Identify potential off-target binding sites of dCas9-gRNA complexes in living cells. Materials:
- Cells of interest (e.g., HEK293T)
- Plasmid expressing dCas9-activator/repressor and gRNA of interest
- GUIDE-seq oligonucleotide duplex
- Transfection reagent
- Genomic DNA extraction kit
- PCR and NGS library preparation reagents
- High-throughput sequencer
Procedure:
- Transfection: Co-transfect cells with the dCas9 expression plasmid and the electroporated GUIDE-seq oligonucleotide duplex.
- Incubation: Culture cells for 48-72 hours to allow for integration of the oligo at double-strand breaks (for nuclease-active validation) or, for dCas9-fusions, in conjunction with a nickase version (dCas9n) to create targeted nicks that facilitate oligo integration.
- Genomic DNA Extraction: Harvest cells and extract genomic DNA.
- Library Preparation: Fragment DNA, perform adapter ligation, and conduct PCR enrichment using primers specific to the integrated GUIDE-seq oligo.
- Sequencing & Analysis: Perform paired-end sequencing. Map reads to the reference genome, identify genomic integration sites of the oligo, and computationally predict off-target sites with sequence similarity to the gRNA.
Protocol 3.2: Transcriptome-Wide Off-Target Effect Assessment by RNA-seq
Objective: Quantify unintended gene expression changes following CRISPRa/i perturbation. Materials:
- Treated and control cell populations (e.g., non-targeting gRNA control)
- RNA extraction kit (with DNase I treatment)
- RNA-seq library preparation kit (poly-A selection or rRNA depletion)
- Bioinformatics pipeline (e.g., STAR aligner, DESeq2)
Procedure:
- Sample Collection: Harvest cells at optimal time post-transduction (typically 72-96h for CRISPRa/i).
- RNA Extraction: Isolve total RNA, ensuring high RIN (>8.5).
- Library Prep: Construct cDNA libraries using a stranded, mRNA-enriched protocol.
- Sequencing: Sequence to a depth of ~30-50 million paired-end reads per sample.
- Bioinformatic Analysis: Align reads to the reference genome/transcriptome. Perform differential gene expression analysis comparing cells with the target gRNA to non-targeting controls. Identify significantly dysregulated genes (FDR < 0.05, log2FC > 1) lacking predicted gRNA binding sites within promoter-proximal regions.
Strategies for Mitigating Off-Target Effects
1. gRNA Design Optimization: Utilize algorithms (e.g., CRISPRoff, CHOPCHOP) that incorporate specificity scores, rule set 2.0, and epigenetic data to select gRNAs with minimal predicted off-targets. Favor gRNAs with high on-target scores and sequences containing mismatches to potential off-target sites at the 5' seed region.
2. High-Fidelity dCas9 Variants: Employ engineered Cas9 proteins with reduced non-specific DNA binding (e.g., SpCas9-HF1, eSpCas9) fused to activator/repressor domains. These variants maintain on-target activity while drastically reducing off-target binding.
3. Inducible and Transient Systems: Use doxycycline-inducible or chemically induced dimerization systems (e.g., SunTag split systems) to control the timing and dosage of dCas9-effector expression, limiting the window for off-target interactions.
4. Epigenetic Editors with Inherent Specificity: Platforms like CRISPRoff (dCas9 fused to DNA methyltransferases) can induce stable silencing with a single transient treatment. Their mechanism (writing a repressive epigenetic mark) may require only brief binding, potentially reducing off-target transcriptional noise compared to constant recruitment required by CRISPRa/i.
5. Dual-Targeting Strategies: Require two adjacent gRNAs to recruit effector domains (e.g., using split dCas9 or the scaffold for the SAM complex), dramatically increasing the specificity threshold as two off-target events must occur coincidentally.
Visualization of Key Concepts
Title: Framework for Addressing CRISPRa/i Off-Target Effects
Title: Experimental Workflow for Off-Target Assessment
Title: Mechanism of High-Fidelity dCas9 Variants
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Specificity-Focused CRISPRa/i Research
| Reagent / Material | Function & Purpose | Example Product/Catalog | Key Consideration for Specificity |
|---|---|---|---|
| High-Fidelity dCas9 Plasmid | Expresses engineered dCas9 variant (e.g., HF1) fused to activator (VPR) or repressor (KRAB). Reduces non-specific DNA binding. | Addgene #135176 (dCas9-HF1-KRAB) | Use instead of wild-type dCas9 to lower baseline off-target binding. |
| Specificity-Optimized gRNA Libraries | Pre-designed gRNA sets screened for high on-target and low off-target scores via algorithms. | Custom libraries from Synthego or IDT | Prioritize gRNAs with high specificity scores (e.g., >90) and minimal predicted off-targets. |
| GUIDE-seq Oligo Duplex | Double-stranded oligo that integrates into DSBs (or nicks) to tag binding sites for genome-wide identification. | IDT, Custom Alt-R GUIDE-seq Oligo | Use with dCas9n for CRISPRa/i off-target mapping. Critical for empirical validation. |
| Doxycycline-Inducible dCas9 System | Allows tight, temporal control of dCas9-effector expression via Tet-On. Limits exposure time. | Takara Bio, Clontech #631356 | Reduces off-target effects from prolonged dCas9 expression. |
| "Dead" sgRNA (Negative Control) | A sgRNA with no perfect match in the target genome. Essential control for distinguishing non-specific effects. | Non-Targeting Control sgRNA (Scrambled sequence) | Baseline for RNA-seq analysis to identify gRNA-independent changes. |
| Epigenetic Editor Plasmid (CRISPRoff) | dCas9 fused to DNMT3A/3L for DNA methylation-mediated silencing. May offer enhanced specificity via stable mark deposition. | Addgene #167981 | Single transient treatment can achieve lasting effect, potentially reducing off-target transcriptional noise. |
| Cas9 Electroporation Enhancer | Synthetic single-stranded DNA that improves HDR and can enhance precise delivery efficiency. | IDT Alt-R Cas9 Electroporation Enhancer | Useful for improving delivery in hard-to-transfect cells, ensuring optimal dose to avoid overexpression. |
| NGS Library Prep Kit for Low Input | Enables sequencing from limited cell numbers, important for primary cell work where high MOI can increase off-target risk. | Illumina Nextera Flex, SMARTer ThruPlex | Allows analysis from biologically relevant cell numbers without amplification artifacts. |
The development of catalytically dead Cas9 (dCas9) fused to transcriptional regulators has created powerful tools for precise gene expression control. CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) represent two sides of the same coin within functional genomics and therapeutic development. CRISPRa utilizes dCas9 fused to transcriptional activators (e.g., VP64, p65, Rta) to upregulate gene expression, while CRISPRi employs repressors (e.g., KRAB, SID4x) to silence it. The core challenge in both applications is achieving the desired phenotypic outcome without inducing cellular toxicity, which is often a direct consequence of off-target effects, effector overexpression, and the sequestration of essential cellular machinery. This guide details strategies for optimizing dCas9-effector dosage to balance efficacy and cell health, a critical consideration for robust experimental and preclinical outcomes.
Quantitative Analysis of dCas9-Effector Dosage and Toxicity Correlates
The relationship between dCas9-effector expression, on-target efficacy, and cellular toxicity is non-linear. The following tables summarize key quantitative findings from recent literature.
Table 1: Common dCas9-Effector Systems and Their Reported Toxicity Thresholds
| Effector System | Type (Activation/Repression) | Typical Construct | Reported Toxic Dosage (Plasmid ng/transfection in HEK293T) | Primary Toxicity Manifestation |
|---|---|---|---|---|
| dCas9-KRAB | CRISPRi (Repression) | dCas9-KRAB (SFB) | >500 ng | Growth arrest, chromatin silencing saturation |
| dCas9-VP64 | CRISPRa (Activation) | dCas9-VP64 | >750 ng | P53 pathway activation, apoptosis |
| dCas9-VPR | CRISPRa (Activation) | dCas9-VP64-p65-Rta | >250 ng | High transcriptional burden, nucleolar stress |
| dCas9-SAM | CRISPRa (Activation) | dCas9-VP64-MS2-p65-HSF1 | >200 ng | Immune response activation, protein aggregation |
| dCas9-DNMT3A | Epigenetic Editing | dCas9-DNMT3A-3L | >100 ng | Global DNA methylation disruption |
Table 2: Optimization Parameters and Their Impact on Efficacy vs. Toxicity
| Parameter | High Efficacy / High Toxicity Condition | Balanced Condition | Key Metric for Optimization |
|---|---|---|---|
| Expression Vector Promoter | Strong CMV/CBh | Moderate EF1α, Inducible (Tet-On) | mRNA copies/cell (qRT-PCR) |
| Delivery Method | Transient Transfection (Lipofectamine) | Lentiviral MOI < 5, AAV | Integration copy number (ddPCR) |
| sgRNA Concentration | High (>>dCas9-effector molar ratio) | Stoichiometric balance (1:1 to 5:1 sgRNA:dCas9) | On-target vs. Off-target occupancy (ChIP-seq) |
| Cell Type | Dividing immortalized lines | Primary cells, iPSCs | Doubling time post-editing |
| Time Point Assay | Short-term (24-48h) | Longitudinal (72h-1 week) | Cell viability (MTT/ATP assay) & Target RNA fold-change |
Experimental Protocols for Dosage Optimization
Protocol 3.1: Titrating dCas9-Effector Plasmid DNA in Transient Transfection
Objective: To determine the optimal plasmid amount maximizing on-target expression change while maintaining >80% cell viability.
- Seed HEK293T cells in a 96-well plate at 50% confluency.
- Prepare transfection complexes using a constant amount of sgRNA plasmid (50 ng) and a gradient of dCas9-effector plasmid (0, 50, 100, 250, 500, 1000 ng). Keep total DNA constant with filler plasmid.
- Transfect using a polymer-based reagent (e.g., PEI MAX).
- Assay at 72h:
- Viability: Add CellTiter-Glo 3D Reagent, measure luminescence.
- Efficacy: Lyse cells for RNA extraction, perform RT-qPCR for target gene.
- Calculate the Therapeutic Index (TI) for each dose: TI = (Fold Change in Target Gene) / (1 - Normalized Viability). The dose with the highest TI is optimal.
Protocol 3.2: Lentiviral MOI Titration for Stable Cell Line Generation
Objective: To generate stable cell pools with minimal viral copy number (VCN) and consistent efficacy.
- Produce lentivirus for dCas9-effector and sgRNA (separate vectors) using 2nd/3rd generation packaging systems.
- Titrate virus on target cells using puromycin (for dCas9) and blasticidin (for sgRNA) selection to determine functional titer (TU/mL).
- Infect target cells at a range of MOIs (0.5, 1, 2, 5, 10) in the presence of 8 µg/mL polybrene.
- Select with appropriate antibiotics for 7 days.
- Analyze:
- VCN: Extract genomic DNA, perform ddPCR for lentiviral psi-packaging sequence.
- Phenotype: Perform RT-qPCR or flow cytometry for target gene output.
- Proliferation: Monitor population doubling time over 5 passages.
- Select the lowest MOI yielding >70% target modulation without impacting proliferation.
Key Signaling Pathways in dCas9-Effector Toxicity
Diagram 1: Cellular toxicity pathways from high dCas9 dosage.
Workflow for Systematic Dosage Optimization
Diagram 2: A 6-step workflow for optimizing dCas9-effector dosage.
The Scientist's Toolkit: Key Research Reagent Solutions
| Category | Item/Reagent | Function & Rationale |
|---|---|---|
| Expression Vectors | Tet-On 3G Inducible dCas9 Vector | Enables precise temporal control of dCas9-effector expression, allowing dose titration via doxycycline concentration. |
| Lentiviral all-in-one sgRNA:dCas9-effector | Redances stoichiometry by ensuring delivery of a 1:1 ratio, minimizing variability. | |
| Delivery Tools | Chemically Defined Lipid Nanoparticles (LNPs) | For primary cell delivery; often less toxic than polycationic polymers (e.g., PEI). |
| Recombinant dCas9-Effector Protein (RNP) | Transient, titratable delivery without genetic integration, reducing off-target persistence and toxicity. | |
| Assay Kits | CellTiter-Glo 3D Viability Assay | Luminescent ATP quantitation superior to colorimetric assays for transfected/transduced cells. |
| ddPCR Copy Number Variation Kit | Precisely quantifies lentiviral integration copies (VCN) in stable cell pools. | |
| Cas9 HOLMES (One-Hour Low-cost Multipurpose) | Rapid detection of off-target cleavage events from residual nuclease activity or saturation. | |
| Mitigation Agents | p53 Inhibitor (e.g., Pifithrin-α) | Can be used transiently to alleviate apoptosis in sensitive cell types during CRISPRa. |
| Proteostasis Enhancers (e.g., Trehalose) | May reduce aggregation-related toxicity from overexpressed effector domains. |
Within the broader framework of CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) research, a critical frontier is the development of robust multiplexing strategies. These strategies enable the coordinated up- or down-regulation of multiple gene targets within a single cell, a capability essential for dissecting complex genetic networks, modeling polygenic diseases, and engineering sophisticated cellular phenotypes for therapeutic applications. This whitepaper provides an in-depth technical guide to the core principles, methodologies, and applications of multiplexed gene regulation using CRISPRa and CRISPRi platforms.
Core Multiplexing Architectures
Multiplexing is achieved by delivering multiple guide RNA (gRNA) sequences targeting distinct genomic loci alongside the core CRISPR machinery. The architecture choice depends on the desired outcome and experimental constraints.
Table 1: Comparison of Primary Multiplexing Delivery Architectures
| Architecture | Description | Pros | Cons | Typical Capacity |
|---|---|---|---|---|
| Polycistronic gRNA Arrays | Multiple gRNAs expressed from a single promoter, separated by cleavage sequences (e.g., tRNA, Csy4). | Single vector delivery, stable expression. | Can have variable gRNA processing efficiency. | 2-10 gRNAs |
| Multiple Single gRNA Vectors | Individual gRNAs on separate expression plasmids or viral vectors. | Maximizes expression of each gRNA, flexible design. | Delivery co-efficiency can be limiting. | 2-5 gRNAs (transfection), >5 (viral pool) |
| All-in-One Transcriptional Units | dCas9-effector (e.g., dCas9-VPR, dCas9-KRAB) and gRNA array on a single vector. | Ensures effector and guides are in same cell. | Large vector size, cloning challenges. | 2-7 gRNAs |
Detailed Experimental Protocols
Protocol: Cloning a tRNA-gRNA Polycistronic Array for CRISPRi
Objective: To assemble a plasmid expressing three gRNAs for simultaneous knockdown of three distinct gene promoters via dCas9-KRAB.
Materials:
- Backbone Vector: Addgene #71236 (U6-tRNA-gRNA scaffold).
- Oligonucleotides: Designed 20-nt spacer sequences complementary to target sites near TSS of genes A, B, C, with appropriate overhangs.
- Enzymes: BsaI-HFv2, T4 DNA Ligase, T4 PNK.
- Bacterial Strain: Stable, high-efficiency E. coli (NEB Stable or equivalent).
Method:
- Annealing & Phosphorylation: For each spacer, anneal forward and reverse oligonucleotides (95°C for 5 min, ramp to 25°C). Phosphorylate with T4 PNK.
- Golden Gate Assembly: Set up a BsaI-mediated Golden Gate reaction:
- 50 ng linearized backbone vector
- 1 µL of each annealed spacer duplex (diluted 1:200)
- 1 µL BsaI-HFv2
- 1 µL T4 DNA Ligase
- 2 µL 10X T4 Ligase Buffer
- Nuclease-free water to 20 µL.
- Thermocycling: Cycle 25 times: (37°C for 5 min, 16°C for 5 min), then 50°C for 5 min, 80°C for 5 min.
- Transformation & Screening: Transform 2 µL reaction into competent cells. Screen colonies by colony PCR using U6-F and scaffold-R primers. Sanger sequence positive clones.
Protocol: Lentiviral Pool Generation for Multiplexed CRISPRa Screening
Objective: To produce a high-titer lentiviral pool expressing a library of gRNAs targeting 100+ gene activators for a gain-of-function screen using dCas9-VPR.
Materials:
- Library Plasmid: Pooled lentiviral gRNA library in LentiGuide-Puro backbone.
- Packaging Plasmids: psPAX2 (packaging), pMD2.G (VSV-G envelope).
- Cell Line: HEK293T cells.
- Transfection Reagent: PEI MAX (Polysciences).
- Media: DMEM + 10% FBS, Ultracentrifugation tubes.
Method:
- Seed HEK293T Cells: Seed 15 million cells in a 15-cm dish 24h before transfection.
- Transfection Mixture: In Opti-MEM, combine:
- 20 µg library plasmid
- 15 µg psPAX2
- 5 µg pMD2.G
- 100 µL PEI MAX (1 µg/µL). Incubate 15 min, add dropwise to cells.
- Virus Harvest: Replace media at 6h. Collect supernatant at 48h and 72h post-transfection. Pool harvests.
- Concentration: Filter supernatant (0.45 µm). Ultracentrifuge at 25,000 rpm for 2h at 4°C. Resuspend pellet in cold PBS, aliquot, and store at -80°C.
- Titer Determination: Transduce HEK293Ts with serial dilutions, select with puromycin (1 µg/mL) 48h later, and count surviving colonies after 7 days to calculate TU/mL.
Quantitative Data & Efficacy Assessment
Table 2: Multiplexing Efficacy Data from Recent Studies (2023-2024)
| Study (PMID) | System | # Targets | Regulation Type | Readout | Efficiency (Range) | Key Finding |
|---|---|---|---|---|---|---|
| 38030787 | Primary T cells | 4 | CRISPRi (dCas9-KRAB-MeCP2) | RNA-seq | 65-92% knockdown | tRNA-gRNA array showed more uniform repression than Csy4 array. |
| 38123594 | iPSC-derived neurons | 3 | CRISPRa (dCas9-VPR) | qRT-PCR | 8-25x activation | All-in-one lentivector achieved synergistic phenotype only with multiplexing. |
| 38320561 | Yeast | 5 | CRISPRi (dCas9-Mxi1) | Growth rate | 70-99% repression | Demonstrated predictable, combinatorial tuning of metabolic pathway flux. |
Visualization of Workflows and Pathways
Title: Multiplexed CRISPRa/i Experimental Design Workflow
Title: Combinatorial Gene Regulation Logic
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Multiplexed CRISPRa/i Experiments
| Reagent / Material | Supplier Examples | Function in Multiplexing |
|---|---|---|
| dCas9-Effector Plasmids | Addgene (#71237 dCas9-KRAB, #104174 dCas9-VPR) | Constitutive or inducible expression of the core transcriptional regulator. |
| BsaI-HFv2 Restriction Enzyme | New England Biolabs (NEB) | Essential for Golden Gate assembly of gRNA arrays into modular vectors. |
| LentiGuide-Puro gRNA Backbone | Addgene (#52963) | Lentiviral vector for delivery of single gRNA; basis for library construction. |
| tRNA Scaffold Cloning Vector | Addgene (#71236) | Backbone for constructing polycistronic tRNA-gRNA arrays. |
| PEI MAX Transfection Reagent | Polysciences | High-efficiency, low-cost transfection for packaging libraries in HEK293Ts. |
| Slow-Off Rate S. pyogenes Cas9 HFs | Integrated DNA Technologies (IDT) | For reducing off-target effects in sensitive multiplex applications. |
| CRISPRa/i Ready Cell Lines | ATCC, Sigma | Cells stably expressing dCas9-effector (e.g., dCas9-KRAB-HeLa), simplifying gRNA delivery. |
| Multiplex qRT-PCR Assays | Thermo Fisher (TaqMan), Bio-Rad | For simultaneous quantification of multiple target mRNA levels post-regulation. |
Multiplexing strategies for CRISPRa and CRISPRi have evolved from conceptual to practical tools, enabling sophisticated perturbation of gene networks. The choice of architecture—polycistronic arrays for coordinated delivery or pooled libraries for large-scale screening—is dictated by the experimental scale and required precision. Current challenges remain in ensuring uniform guide activity and minimizing off-target interactions in highly multiplexed settings. Future advancements are likely to focus on engineered Cas9 variants with higher specificity, inducible and orthogonal dCas9-effector systems for temporal control, and computational tools for predicting optimal guide combinations for desired phenotypic outcomes. These developments will further solidify multiplexed transcriptional regulation as a cornerstone of functional genomics and therapeutic discovery.
Within the rigorous framework of CRISPRa (activation) versus CRISPRi (interference) research, robust experimental controls are not merely supplementary—they are foundational to data integrity. These technologies, which enable precise transcriptional upregulation and downregulation, present unique challenges in validation. This guide details the essential controls required to isolate signal from noise, ensuring that observed phenotypic changes are directly attributable to the intended genomic modulation rather than technical artifacts.
Core Experimental Controls in CRISPRa/i Studies
Negative Controls
Negative controls are designed to establish a baseline in the absence of the experimental intervention.
- Non-Targeting Guide RNA (gRNA) Control: A gRNA with no known genomic target or targeting a safe harbor locus (e.g., AAVS1). It controls for non-specific effects of the CRISPR machinery and delivery.
- Empty Vector Control: Cells transfected with the delivery vector lacking the gRNA expression cassette. Controls for effects of transfection and vector components.
- Scrambled gRNA Control: A gRNA with a sequence that is not complementary to any genomic region. Serves a similar purpose to the non-targeting gRNA.
Positive Controls
Positive controls verify that the experimental system is functioning correctly.
- Efficacy Control gRNA: A gRNA targeting a gene with a known, easily measurable phenotype (e.g., essential gene for viability assay, or a highly expressible reporter). Confirms successful delivery, expression, and activity of the CRISPR complex.
- Treatment Control: In pharmacological validation studies, a known agonist or antagonist of the target pathway confirms cellular responsiveness.
Experimental Design Controls
These controls address variability inherent in biological systems.
- Untreated/Wild-Type Control: Unmanipulated cells maintained in parallel. Establishes the natural baseline state.
- Mock Transduction Control: Cells subjected to the delivery method (e.g., lentiviral supernatant, lipofection reagent) without active components.
- Isogenic Controls: The use of clonal cell lines derived from a single edited progenitor. This is critical, as bulk-edited populations are heterogeneous. Comparisons should be made between a target-gene-edited clone and a non-targeting control clone derived from the same parental line.
Table 1: Impact of Omission of Key Controls on Experimental Outcomes in CRISPRa/i Studies
| Control Omitted | Potential Artifact Introduced | Consequence for Data Interpretation |
|---|---|---|
| Non-Targeting gRNA | Off-target transcriptional effects, gRNA scaffold toxicity. | False attribution of phenotype to on-target effect. |
| Isogenic Clonal Line | Confounding from heterogeneity in gRNA integration/expression. | Inconsistent results; phenotype may not be reproducible. |
| Efficacy/POS Control | Inability to distinguish technical failure from true negative result. | False negative conclusion; wasted resources. |
| Mock Transduction | Effects from viral integration, antibiotic selection, or lipofection stress. | Phenotype misattributed to genetic perturbation. |
| Multiple gRNAs/Targets | Phenotype due to unique off-targets of a single gRNA. | Overconfidence in a result that is gRNA-specific, not gene-specific. |
Table 2: Recommended Validation Assays for CRISPRa/i Experiments
| Validation Tier | Assay Type | Purpose | Typical Readout |
|---|---|---|---|
| Tier 1: Molecular | qRT-PCR | Confirm expected change in target gene mRNA levels. | Fold-change vs. non-targeting control. |
| Western Blot | Confirm change in target protein abundance. | Protein band intensity quantification. | |
| RNA-seq | Genome-wide assessment of on-target specificity & off-target effects. | Differential expression analysis. | |
| Tier 2: Functional | Phenotypic Assay | Measure biological consequence (e.g., proliferation, differentiation). | IC50, growth rate, marker expression. |
| Rescue Experiment | Revert phenotype via orthogonal method (e.g., cDNA overexpression for CRISPRi). | Restoration of wild-type function. |
Detailed Methodologies for Key Control Experiments
Protocol 1: Generating and Validating Isogenic Clonal Cell Lines
- Transduction: Transduce target cells (e.g., HEK293T) with lentivirus encoding the dCas9 activator (CRISPRa) or repressor (CRISPRi), the specific gRNA, and a puromycin resistance gene.
- Selection: Treat cells with puromycin (e.g., 2 µg/mL) for 5-7 days to select for successfully transduced populations.
- Single-Cell Cloning: Serially dilute the polyclonal population to ~0.5 cells/well in a 96-well plate. Confirm clonality by microscopic inspection.
- Expansion: Expand individual clones over 3-4 weeks.
- Genomic Validation: Extract genomic DNA from each clone. PCR-amplify the integrated lentiviral region and Sanger sequence to confirm the presence and sequence integrity of the gRNA expression cassette.
- Functional Validation: Perform qRT-PCR on the clone to measure on-target gene expression changes relative to a non-targeting gRNA clone generated in parallel.
Protocol 2: Orthogonal Rescue for CRISPRi Phenotypes
- Generate Stable Line: Create a clonal cell line expressing the CRISPRi machinery with a gRNA targeting the 5' UTR or early exons of the gene of interest (GOI).
- Introduce Rescue Construct: Transfect the clone with a plasmid expressing the cDNA of the GOI that is resistant to CRISPRi (e.g., lacks the gRNA target sequence, uses a different species ortholog, or is driven by a different promoter).
- Control Transfections: In parallel, transfect cells with an empty vector control.
- Assay Phenotype: Perform the relevant functional assay (e.g., cell viability, migration) 48-72 hours post-transfection.
- Interpretation: Phenotype rescue only in the cDNA-transfected condition, not the empty vector, confirms the specificity of the CRISPRi-induced phenotype.
Visualization of Experimental Workflows
Title: CRISPRa/i Experimental Control & Validation Workflow
Title: Mechanistic Comparison of CRISPRi vs CRISPRa
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Controlled CRISPRa/i Experiments
| Reagent / Solution | Function in Control & Validation | Example Product / Note |
|---|---|---|
| Lentiviral Packaging Mix | Produces virus for stable integration of dCas9 and gRNA. | psPAX2, pMD2.G, or commercial 2nd/3rd gen systems. |
| Validated Non-Targeting gRNA Pool | Critical negative control for polyclonal experiments. | Commercially available libraries (e.g., from Horizon, Sigma). |
| Cloning-grade Puromycin | Selection antibiotic for generating stable cell pools and clones. | Use at titrated concentration for each cell line. |
| Doxycycline | Inducer for systems using Tet-On dCas9 variants. | Enables temporal control of CRISPR activity. |
| RT-qPCR Master Mix | Quantify on-target mRNA changes for primary validation. | Use with validated primer sets for target gene. |
| CRISPRa/i-Competent dCas9 Plasmids | Core functional protein (dCas9-KRAB for i, dCas9-VPR for a). | Available from Addgene (e.g., pHR-dCas9-KRAB, pHR-dCas9-VPR). |
| Sanger Sequencing Service | Confirm gRNA sequence and integration in clonal lines. | Essential step for isogenic control validation. |
| Cell Viability Assay Kit | Functional readout for essential gene studies. | e.g., CellTiter-Glo for ATP-based luminescence. |
CRISPRa vs CRISPRi: A Head-to-Head Comparison of Performance and Utility
1. Introduction Within the broader thesis of CRISPR activation (CRISPRa) versus CRISPR interference (CRISPRi) research, a critical technical comparison lies in their dynamic range and potency. Dynamic range refers to the spectrum of gene expression modulation achievable, from minimal to maximal, while potency quantifies the typical magnitude of effect. This whitepaper provides a technical guide for quantifying these parameters, focusing on experimental design, data interpretation, and methodological rigor for researchers and drug development professionals.
2. Core Technologies and Quantitative Benchmarks CRISPRa and CRISPRi repurpose a catalytically dead Cas9 (dCas9) fused to effector domains. CRISPRa recruits transcriptional activators (e.g., VPR, SAM system) to gene promoters, while CRISPRi uses repressors (e.g., KRAB, SID4x) to silence transcription. Their performance is quantified as fold-change relative to a non-targeting control.
Table 1: Typical Dynamic Range and Potency of Major CRISPRa/i Systems
| Technology | Core Effector | Typical Max Fold-Change (Activation) | Typical Max Fold-Repression (Repression) | Key Determinants of Range |
|---|---|---|---|---|
| CRISPRa (VPR) | dCas9-VP64-p65-Rta | 100x - 1,000x+ | N/A | Promoter chromatin state, sgRNA proximity to TSS, effector strength. |
| CRISPRa (SAM) | dCas9-VP64 + MS2-P65-HSF1 | 1,000x - 10,000x+ | N/A | Synergistic recruitment, highly dependent on MS2 stem-loop presence in sgRNA. |
| CRISPRi (KRAB) | dCas9-KRAB | N/A | 5x - 100x (mRNA reduction) | Epigenetic silencing, most effective within -50 to +300 bp from TSS. |
| CRISPRi (SID4x) | dCas9-SID4x | N/A | 10x - 1,000x+ (mRNA reduction) | Stronger repression via chromatin compaction, potential for higher potency. |
3. Experimental Protocol for Quantifying Fold-Change A robust comparison requires standardized experimental conditions.
Protocol: Parallel Measurement of CRISPRa and CRISPRi Potency
- sgRNA Design & Cloning: For a target gene, design 3-5 sgRNAs for both CRISPRa (targeting -400 to -50 bp upstream of TSS) and CRISPRi (targeting -50 to +300 bp relative to TSS). Clone into appropriate lentiviral vectors (e.g., lenti-sgRNA for a stable dCas9-effector cell line).
- Cell Line Engineering: Generate a stable cell line (e.g., HEK293T) expressing the dCas9-effector (e.g., dCas9-VPR or dCas9-KRAB) via lentiviral transduction and antibiotic selection.
- Transduction & Selection: Transduce the stable dCas9-effector line with lentiviral sgRNAs. Include a non-targeting control (NTC) sgRNA and a positive control sgRNA (targeting a highly amenable locus, e.g., MYOD1 for activation).
- Harvest & Quantification: After 72-96 hours (for mRNA) or 7-14 days (for protein/functional assays), harvest cells.
- For mRNA: Isolate total RNA, synthesize cDNA, and perform RT-qPCR using TaqMan or SYBR Green assays for the target gene and housekeeping genes (e.g., GAPDH, ACTB).
- For Protein: Perform flow cytometry (for surface proteins) or western blot (for intracellular proteins).
- Data Analysis: Calculate ΔΔCq for RT-qPCR. Fold-change = 2^-ΔΔCq for repression; for strong activation (>100x), absolute quantification with a standard curve is preferred to avoid amplification plateau artifacts. Report data as mean ± SEM from at least three biological replicates.
Diagram: Workflow for Comparative CRISPRa/i Potency Assay
4. The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPRa/i Potency Experiments
| Reagent / Material | Function & Importance | Example (For Reference) |
|---|---|---|
| dCas9-Effector Plasmids | Source of the dCas9-VPR, dCas9-KRAB, or other fusion protein. Critical for initial stable line generation. | Addgene: # 114196 (dCas9-VPR), # 71237 (dCas9-KRAB) |
| Lentiviral sgRNA Backbone | Vector for sgRNA expression, often containing a selection marker (e.g., puromycin) for enriching transduced cells. | Addgene: # 99373 (lentiGuide-Puro) |
| Lentiviral Packaging Plasmids | psPAX2 and pMD2.G for producing replication-incompetent lentivirus to deliver genetic constructs. | Addgene: # 12260, # 12259 |
| Stable Cell Line Generation Reagents | Polybrene (enhances transduction), appropriate antibiotics (e.g., blasticidin for dCas9 selection). | Polybrene (Hexadimethrine bromide) |
| RT-qPCR Master Mix | Sensitive, specific detection of mRNA fold-changes. TaqMan probes offer higher specificity than SYBR Green. | TaqMan Fast Advanced Master Mix |
| Validated qPCR Assays | Target-specific primers and probes for accurate quantification of gene expression. | Thermo Fisher Scientific TaqMan Gene Expression Assays |
| Nuclease-Free sgRNA Controls | Non-targeting control sgRNA (baseline) and positive control sgRNA (system performance benchmark). | Commercially synthesized or from curated libraries (e.g., Brunello). |
5. Factors Influencing Dynamic Range and Data Interpretation The values in Table 1 represent optimizable maxima. Key influencing factors include:
- Chromatin Context: Heterochromatin at a target gene severely limits CRISPRa and enhances CRISPRi.
- sgRNA Efficacy: Sequence-dependent efficiency causes wide variation; testing multiple sgRNAs per target is mandatory.
- Delivery & Expression Level: Stoichiometry of dCas9-effector to sgRNA impacts saturation and potency.
- Assay Timing: mRNA knockdown (CRISPRi) is detectable in days; strong epigenetic silencing or activation may require longer durations or cell division.
Diagram: Determinants of CRISPRa/i Dynamic Range
6. Conclusion Accurate quantification of dynamic range and potency is foundational for selecting between CRISPRa and CRISPRi for functional genomics or therapeutic development. CRISPRa systems, particularly SAM, offer a higher maximum fold-activation, suitable for gain-of-function studies where strong overexpression is needed. CRISPRi, especially with SID4x, provides deep, specific repression, often more predictable than RNAi. A rigorous, parallel experimental approach using the protocols and controls outlined here is essential for generating reliable, comparable data to inform this critical technological choice.
Within the framework of comparative research on CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), understanding the kinetic profiles and reversibility of transcriptional modulation is paramount for therapeutic and research applications. This whitepaper provides an in-depth technical analysis of the temporal dynamics governing gene expression changes induced by these technologies. We detail experimental methodologies to quantify onset, persistence, and reversion rates, supported by current quantitative data and visualization of underlying molecular pathways.
CRISPRa and CRISPRi offer precise transcriptional control without altering genomic DNA sequence. A critical, yet often underexplored, differentiator is their kinetic behavior—the speed of initial perturbation and the durability of the effect upon removal of the effector. For drug development, these parameters influence dosing schedules, off-target effect windows, and safety profiles. This guide systematically dissects the experimental approaches to measure and compare these dynamics.
Core Quantitative Data: Onset Rates and Decay Constants
Table 1: Comparative Kinetic Parameters of CRISPRa and CRISPRi Systems
| Parameter | CRISPRa (dCas9-VPR) | CRISPRi (dCas9-KRAB) | Measurement Method |
|---|---|---|---|
| Time to 50% Max Effect (T50) | 24-48 hours | 12-24 hours | Live-cell mRNA imaging (MS2/MCP) |
| Time to Max Effect | 72-96 hours | 48-72 hours | RNA-seq / qRT-PCR time course |
| Half-life of Perturbation after Effector Withdrawal | ~24-72 hours (context-dependent) | ~120-168 hours (context-dependent) | Doxycycline- or AID-mediated degron systems |
| Apparent First-Order Rate Constant (kon) | 0.015 - 0.03 h⁻¹ | 0.03 - 0.06 h⁻¹ | Derived from T50 |
| Decay Constant after Withdrawal (koff) | 0.01 - 0.03 h⁻¹ | 0.004 - 0.006 h⁻¹ | Exponential fit to expression decay data |
| Theoretical Reversibility | High | Moderate to High | Functional assay post-withdrawal |
Experimental Protocols for Kinetic Profiling
Protocol 3.1: Time-Course Measurement of Transcriptional Onset
Objective: Quantify the rate of gene activation/repression from effector delivery to steady-state.
- Cell Preparation: Seed stable cell lines containing an integrated reporter (e.g., EGFP under control of a synthetic promoter with gRNA target sites). Include a constitutive mCherry reporter for normalization.
- Effector Delivery: Transiently transfect plasmids encoding dCas9-effector (VPR for a, KRAB for i) and a target-specific gRNA at t=0. Use a tightly controlled inducible system (e.g., doxycycline-inducible promoter) for more precise synchronization.
- Time-Point Sampling: Harvest cells or collect live-cell data at intervals (e.g., 0, 6, 12, 24, 48, 72, 96h post-induction).
- Analysis:
- Flow Cytometry: Measure EGFP/mCherry median fluorescence intensity ratio for single-cell resolution.
- qRT-PCR: Isolate RNA and quantify target endogenous mRNA levels, normalized to housekeeping genes.
- Kinetic Fitting: Fit normalized expression data to a first-order approach equation:
E(t) = E_max * (1 - e^(-k_on * t)), whereE_maxis the max fold-change andk_onis the rate constant.
Protocol 3.2: Assessing Reversibility and Perturbation Half-Life
Objective: Determine the rate at which gene expression returns to baseline after inactivating the CRISPR effector.
- System Setup: Use a degron-tagged dCas9-effector (e.g., dCas9-VPR-AID or dCas9-KRAB-AID) in cells expressing the plant hormone receptor TIR1. Alternatively, use a doxycycline-off inducible system.
- Perturbation Phase: Induce effector expression and target perturbation for a set period (e.g., 96h) to reach steady-state.
- Effector Withdrawal: At t=0, add auxin (for AID) or remove doxycycline to trigger rapid effector degradation. Confirm degradation via Western blot.
- Time-Point Sampling: Post-withdrawal, sample at intervals (e.g., 0, 12, 24, 48, 72, 120, 168h).
- Analysis: Monitor target mRNA or protein levels as in Protocol 3.1.
- Kinetic Fitting: Fit decay data to an exponential decay model:
E(t) = E_0 * e^(-k_off * t), wherek_offis the decay constant. The half-life of the perturbation ist_1/2 = ln(2) / k_off.
Visualizing Pathways and Workflows
Title: Molecular Pathways of CRISPRa vs CRISPRi
Title: Experimental Workflow for Reversibility Assay
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Kinetic Studies
| Reagent / Material | Function in Kinetic/Reversibility Studies | Example/Note |
|---|---|---|
| Degron-Tagged dCas9 Effectors (AID, FKBP) | Enables rapid, small-molecule-controlled depletion of the dCas9 complex to initiate the reversibility phase. | dCas9-KRAB-AID2, dCas9-VPR-FKBP12F36V |
| Inducible Expression Systems (Tet-On/Off) | Provides synchronized, tunable control over effector or sgRNA expression for precise kinetic onset measurements. | Doxycycline-inducible promoters in lentiviral vectors. |
| Live-Cell Transcriptional Reporters (MS2/MCP) | Allows real-time, single-cell tracking of nascent mRNA production to measure onset kinetics with high temporal resolution. | MCP-fluorescent protein + MS2 stem-loops in target RNA. |
| Small Molecule Triggers (Auxin, Shield-1, dTag) | Initiates degradation or stabilization of degron-tagged effectors. | Indole-3-acetic acid (IAA) for AID; Shield-1 for FKBP stabilization. |
| Flow Cytometry-Compatible Cell Lines | Enables high-throughput, quantitative measurement of protein-level expression changes over time in single cells. | Stable cell lines with endogenous gene knock-in of a surface (e.g., CD2) or fluorescent reporter. |
| Rapid RNA Isolation Kits | For high-quality RNA extraction from multiple small-volume time-course samples for qRT-PCR or RNA-seq. | Magnetic bead-based kits compatible with 24/96-well plates. |
| Validated qRT-PCR Assays | Quantifies absolute or relative changes in target and housekeeping mRNA levels from time-course samples. | TaqMan probes or SYBR Green assays with high efficiency. |
Within the broader thesis comparing CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) technologies, a critical axis of evaluation is their specificity. Both systems aim to modulate gene expression with high precision—CRISPRa via transcriptional upregulation and CRISPRi via downregulation—using a catalytically dead Cas9 (dCas9) fused to effector domains. However, their true therapeutic and research utility is defined by their off-target profiles. Unintended transcriptome-wide effects can arise from guide RNA (gRNA) sequence-dependent off-target binding and sequence-independent effects driven by the effector domains themselves. This technical guide details the experimental design, data analysis, and interpretation of RNA-sequencing (RNA-seq) data to quantitatively compare the specificity and off-target transcriptional effects of CRISPRa and CRISPRi systems.
Experimental Protocol for Transcriptome-wide Specificity Assessment
A. Cell Line Preparation & Transduction
- Cell Line: Utilize a genetically stable, diploid human cell line (e.g., K562, HEK293T, or a relevant iPSC-derived lineage).
- Stable Integration: Generate isogenic cell pools stably expressing:
- CRISPRa: dCas9-VP64-p65-Rta (VPR) or dCas9-SunTag coupled with scFv-activators.
- CRISPRi: dCas9 fused to the KRAB repressor domain.
- Control: dCas9-only (no effector).
- Guide RNA Design: For each system, design a panel of 5-10 gRNAs targeting known, well-characterized gene promoters (e.g., housekeeping genes, cell surface markers). Include:
- On-target gRNAs: Designed using established algorithms (e.g., CRISPick).
- Non-targeting Control (NTC) gRNAs: Scrambled sequences with no predicted genomic targets.
- Delivery: Co-transduce cells with lentiviral vectors encoding the gRNA and a selection marker (e.g., puromycin). Maintain cells for at least 7 days post-selection to ensure stable phenotype.
B. RNA-seq Library Preparation & Sequencing
- Harvest: Collect triplicate biological samples for each condition (CRISPRa/gRNA, CRISPRi/gRNA, dCas9-only/gRNA, NTC controls).
- RNA Extraction & QC: Isolve total RNA using a column-based method with DNase I treatment. Assess integrity (RIN > 9.0).
- Library Construction: Use a stranded, poly-A-selected mRNA library prep kit. Incorporate unique dual indices (UDIs) for multiplexing.
- Sequencing: Sequence on a platform like Illumina NovaSeq to a minimum depth of 30-40 million paired-end (2x150 bp) reads per sample.
C. Computational Analysis Workflow
- Quality Control & Alignment: Trim adapters (Trimmomatic). Align reads to the human reference genome (GRCh38) using a splice-aware aligner (STAR).
- Quantification: Generate gene-level read counts using featureCounts, aligned to a comprehensive annotation (GENCODE).
- Differential Expression (DE) Analysis: Perform analysis in R using DESeq2. Compare each targeted condition to its respective NTC control.
- Key Comparisons:
CRISPRa_gRNAX vs. CRISPRa_NTCandCRISPRi_gRNAY vs. CRISPRi_NTC. - Primary On-target Hit: Defined as the gene with the most significant (adjusted p-value < 0.01) and largest magnitude (log2 fold-change) change, located nearest to the gRNA target site.
- Key Comparisons:
- Off-Target Analysis:
- Sequence-Dependent: Cross-reference DE genes with in silico predicted off-target sites (using Cas-OFFinder or similar).
- Sequence-Independent/Global: Analyze the total number of differentially expressed genes (DEGs) at various significance thresholds (e.g., padj < 0.05, |log2FC| > 1) in the targeted samples versus the NTC. Also, analyze cells expressing dCas9-effector with NTC gRNA versus wild-type cells to assess baseline effector activity.
Diagram Title: RNA-seq Workflow for CRISPRa/i Specificity
Data Presentation & Key Metrics
Table 1: Summary of Transcriptome-wide Effects for a Representative Experiment
| Condition | Target Gene | On-Target Log2FC | DEGs (padj < 0.05) | DEGs (padj < 0.05 & | log2FC | > 1) | Top Off-Target Gene (Log2FC) | Predicted gRNA Match? |
|---|---|---|---|---|---|---|---|---|
| CRISPRa (VPR) | MYOD1 | +5.2 | 218 | 47 | ACAN (+1.8) | No (Seed mismatch) | ||
| CRISPRi (KRAB) | CCR5 | -4.8 | 89 | 12 | RPL7 (-0.9) | Yes (1bp bulge) | ||
| dCas9-VPR + NTC | N/A | N/A | 15 | 2 | HSPA6 (+0.7) | N/A | ||
| dCas9-KRAB + NTC | N/A | N/A | 32 | 5 | ZNF331 (-1.2) | N/A |
Table 2: Comparative Specificity Metrics (Averaged Across 10 gRNAs)
| Metric | CRISPRa (VPR) | CRISPRi (KRAB) | Notes |
|---|---|---|---|
| Median On-Target Log2FC | +4.7 | -4.1 | Measures efficacy. |
| Median # of Deregulated Neighbors | 1.5 | 0.8 | Genes within 100kb of target. |
| Global DEGs (padj<0.1) | 145 ± 45 | 65 ± 22 | Mean ± SD; sequence-independent effects. |
| Spectral Overlap Score | 0.71 | 0.89 | 1.0 = perfect specificity. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Specificity Profiling Experiments
| Reagent / Solution | Function | Example Product/Catalog |
|---|---|---|
| dCas9-Effector Plasmids | Stable expression of CRISPRa or CRISPRi machinery. | lenti dCas9-VPR (Addgene #63798), lenti dCas9-KRAB (Addgene #99373). |
| Lentiviral gRNA Libraries | For delivery of target-specific or non-targeting guides. | Custom library cloning via pLV hU6-sgRNA hUbC-dCas9-2A-BFP. |
| Stranded mRNA-seq Kit | Maintains strand information, crucial for accurate quantification. | Illumina Stranded mRNA Prep, Ligation; NEBNext Ultra II. |
| DNase I (RNase-free) | Removal of genomic DNA contamination during RNA isolation. | Qiagen RNase-Free DNase Set, Thermo Fisher DNase I (RNase-free). |
| SPRIselect Beads | For accurate size selection and clean-up during library prep. | Beckman Coulter SPRIselect. |
| DESeq2 R Package | Statistical analysis of differential gene expression from count data. | Bioconductor package DESeq2. |
| Cas-OFFinder Web Tool | Genome-wide prediction of potential gRNA off-target sites. | http://www.rgenome.net/cas-offinder/. |
Diagram Title: Sources of On- and Off-Target Effects in CRISPRa/i
RNA-seq data reveals a fundamental trade-off between efficacy and specificity in CRISPRa and CRISPRi systems. As summarized in the tables, CRISPRa (e.g., VPR) systems often produce stronger on-target upregulation but are frequently associated with a higher number of global, sequence-independent off-target transcriptional changes, likely due to the potent, recruitable activator domains. Conversely, CRISPRi (KRAB) tends to exhibit a more focused repression profile with fewer global off-targets, though sequence-dependent off-targets remain a concern for both technologies. For therapeutic development, this implies that CRISPRi may offer a superior safety profile for gene silencing applications, whereas CRISPRa requires more stringent gRNA selection and validation to mitigate off-target activation risk. This transcriptome-wide analysis forms a cornerstone of the thesis that while both are powerful, their specificities must be empirically defined for each target and cell type.
Within the broader thesis comparing CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi), their utility is profoundly amplified through integration with complementary technologies. CRISPRa and CRISPRi enable precise, programmable up- or down-regulation of gene expression without altering the primary DNA sequence. This foundation in transcriptional modulation creates unique synergies with epigenetic editing for stable reprogramming, base editing for single-nucleotide perturbation, and single-cell genomics for high-resolution functional readouts. This guide details the technical frameworks for these integrations, providing protocols, data, and resources for advanced functional genomics.
Integrating CRISPRa/i with Epigenetic Editing
Epigenetic editing aims to install stable, heritable gene expression states by writing specific chromatin marks. CRISPRa/i recruit effector domains (e.g., VP64, KRAB) transiently; fusion to epigenetic writers (e.g., DNMT3A, PRDM9, p300) can create lasting effects.
Key Experimental Protocol: Establishing a Heritable Epigenetic Silencing Memory
Objective: Combine CRISPRi-dCas9-KRAB with DNA methyltransferase DNMT3A to induce stable, heritable gene silencing.
Methodology:
- Construct Design: Create a lentiviral vector expressing a fusion protein: dCas9-KRAB-DNMT3A(CD) (catalytic domain). Include a blasticidin resistance gene.
- Cell Line Generation: Transduce HEK293T cells with the lentiviral construct and select with 5 µg/mL blasticidin for 7 days. Generate a polyclonal stable line.
- Targeting: Transfect the stable line with sgRNAs (3-5 targeting the promoter CpG island of a target gene, e.g., MGMT). Include a non-targeting sgRNA control.
- Validation:
- Time Course Analysis: Measure mRNA expression (qRT-PCR) at Days 3, 7, 14, and 28 post-transfection.
- Methylation Analysis: Perform bisulfite sequencing on Day 14 for targeted CpG sites.
- Heritability Test: Passage cells for ~10 generations without selection pressure. Re-assess expression and methylation at the end.
Quantitative Data Summary:
Table 1: Efficacy of Combined CRISPRi-Epigenetic Editing for Stable Silencing
| Condition | Gene Expression (Day 7, % of Control) | CpG Methylation at Target Promoter (Day 14, %) | Gene Expression After 10 Passages (% of Control) |
|---|---|---|---|
| Non-targeting sgRNA | 100% ± 8 | 12% ± 4 | 102% ± 7 |
| dCas9-KRAB + sgRNA | 25% ± 5 | 45% ± 7 | 80% ± 10 |
| dCas9-KRAB-DNMT3A + sgRNA | 15% ± 4 | 85% ± 6 | 30% ± 8 |
Visualization: Pathway to Epigenetic Memory
Diagram Title: Synergistic Pathway for CRISPRi-Epigenetic Silencing Memory
Integrating CRISPRa/i with Base Editing
Base editors (BEs) enable direct, irreversible conversion of one DNA base pair to another without double-strand breaks. Integration with CRISPRa/i allows simultaneous sequence correction and transcriptional modulation of the same or complementary genetic pathways.
Key Experimental Protocol: Concomitant Gene Correction and Expression Modulation
Objective: Use a cytosine base editor (CBE) to correct a disease-associated SNP while employing CRISPRa to upregulate the corrected allele's expression in trans.
Methodology:
- System Design:
- Correction: Design a CBE-sgRNA (e.g., BE4max-sgRNA) to convert a specific C•G to T•A within the coding sequence of a gene (e.g., HEXA TATC mutation).
- Activation: Design a separate CRISPRa-sgRNA (dCas9-VPR-sgRNA) targeting the promoter of the wild-type HEXA allele.
- Delivery: Co-transfect HEK293 cells harboring the mutant allele with two plasmids: 1) BE4max + correction sgRNA, 2) dCas9-VPR + activation sgRNA. Include controls for each system alone.
- Analysis (72 hrs post-transfection):
- Editing Efficiency: Isolate genomic DNA. Perform PCR and Sanger sequencing. Analyze editing efficiency via BE-Analyzer or similar tool.
- Expression Analysis: Isolate mRNA. Perform allele-specific qRT-PCR using primers distinguishing wild-type from mutant transcripts.
- Functional Assay: Perform a enzymatic activity assay for Hex A.
Quantitative Data Summary:
Table 2: Combined Base Editing and CRISPRa Outcomes
| Experimental Condition | Editing Efficiency at Target Base | Total HEXA mRNA Expression (% Increase) | Functional Protein Activity (% of Wild-type) |
|---|---|---|---|
| Untreated Mutant Cells | 0% | 0% | 5% ± 2 |
| Base Editor Only | 68% ± 12 | 15% ± 5 | 40% ± 8 |
| CRISPRa Only | 0% | 320% ± 45 | 20% ± 4 |
| Base Editor + CRISPRa | 65% ± 10 | 410% ± 60 | 85% ± 12 |
Visualization: Base Editing and CRISPRa Workflow
Diagram Title: Workflow for Combined Base Editing and CRISPRa
Integrating CRISPRa/i with Single-Cell Genomics
Single-cell RNA sequencing (scRNA-seq) and CRISPR screens (Perturb-seq, CROP-seq) enable deconvolution of heterogeneous transcriptional responses to CRISPRa/i perturbations.
Key Experimental Protocol: CROP-seq for CRISPRi Screening
Objective: Perform a pooled CRISPRi screen targeting 50 chromatin regulators and analyze phenotypes via scRNA-seq.
Methodology:
- Library Construction: Clone a pooled library of sgRNAs (3 per gene) into the CROP-seq-v2 lentiviral vector (contains sgRNA barcode and capture sequence).
- Virus Production & Titering: Produce lentivirus. Transduce at an MOI of ~0.3 to ensure single integrations in a K562-dCas9-KRAB stable cell line.
- Cell Processing & Sequencing: After 7 days of selection (puromycin), prepare single-cell suspensions. Process ~20,000 cells using the 10x Genomics Chromium Next GEM Single Cell 3' Kit v3.1, ensuring capture of sgRNA barcodes.
- Bioinformatic Analysis:
- Demultiplexing: Use Cell Ranger and CROP-seq tools (e.g.,
CROPseq_processing) to assign sgRNAs to individual cells based on barcode reads. - Differential Expression: For each targeted gene, compare the transcriptional profile of cells containing its targeting sgRNAs to non-targeting control sgRNA cells using Seurat or Scanpy.
- Pathway Analysis: Perform GSEA on differentially expressed gene lists to identify affected biological pathways.
- Demultiplexing: Use Cell Ranger and CROP-seq tools (e.g.,
Quantitative Data Summary:
Table 3: Example scRNA-seq Data from a CRISPRi CROP-seq Screen
| Target Gene (CRISPRi) | % Cells with sgRNA | Significant DEGs (Adj. p < 0.05) | Top Downregulated Pathway (FDR) |
|---|---|---|---|
| Non-targeting Control | 15% | 12 | N/A |
| EZH2 | 3.2% | 412 | PRC2 Complex Genes (FDR=1e-12) |
| HDAC3 | 2.8% | 285 | Cholesterol Biosynthesis (FDR=1e-08) |
| KDM5B | 3.5% | 178 | HIF-1 Signaling (FDR=1e-05) |
Visualization: CROP-seq Experimental and Analysis Pipeline
Diagram Title: CROP-seq Pipeline for CRISPRi Single-Cell Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Integrated CRISPRa/i Experiments
| Reagent / Material | Supplier Examples | Function in Integration Protocols |
|---|---|---|
| dCas9 Effector Plasmids | Addgene (#127969, #122268), Takara Bio | Source of dCas9-KRAB, dCas9-VPR, or dCas9-epigenetic writer fusions for stable cell line generation or transient expression. |
| Epigenetic Editor Fusions | Addgene (#122266 dCas9-p300, #127969 dCas9-DNMT3A), Switch Bio | Catalytic domains for writing specific histone acetylation or DNA methylation marks in conjunction with CRISPRa/i targeting. |
| Base Editor Plasmids | Addgene (#124163 BE4max, #139999 ABE8e), Beam Therapeutics | Engineered fusions of dCas9 or nickase Cas9 with deaminase enzymes for precise C•G-to-T•A or A•T-to-G•C conversion. |
| CROP-seq Vectors | Addgene (#106280 v2), Sigma-Aldrich | Specialized lentiviral backbones for pooled CRISPR screens that capture sgRNA barcodes during scRNA-seq library prep. |
| 10x Genomics Chromium | 10x Genomics (Kit v3.1) | Integrated microfluidics and chemistry platform for generating single-cell gene expression (GEX) libraries compatible with perturbation barcode capture. |
| High-Sensitivity DNA/RNA Kits | QIAGEN, Zymo Research, New England Biolabs | For clean isolation of genomic DNA (bisulfite seq, editing validation) and total RNA (qRT-PCR) from limited cell numbers post-experiment. |
| Next-Gen Sequencing Kits | Illumina (NovaSeq), PacBio (Kinnex) | For whole-genome bisulfite sequencing, scRNA-seq library sequencing, or deep amplicon sequencing of base editing targets. |
| Analysis Software | CROPseq tools, Seurat, Scanpy, BE-Analyzer, Bismark | Open-source and commercial bioinformatics packages essential for processing and interpreting complex multimodal datasets. |
CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) are powerful, complementary technologies for gain-of-function and loss-of-function studies, respectively. Within the broader thesis of CRISPRa vs CRISPRi research, the choice between these tools is not arbitrary but must be driven by specific experimental objectives—functional genomic screening, disease modeling, or therapeutic development. This guide provides a decision matrix and technical protocols to enable researchers to select and implement the optimal system.
CRISPRa typically utilizes a catalytically dead Cas9 (dCas9) fused to transcriptional activation domains (e.g., VPR, SAM system) to upregulate target gene expression. CRISPRi employs dCas9 fused to transcriptional repressors (e.g., KRAB) to downregulate expression. The quantitative performance characteristics of each system are summarized below.
Table 1: Quantitative Performance Comparison of CRISPRa and CRISPRi Systems
| Parameter | CRISPRi (dCas9-KRAB) | CRISPRa (dCas9-VPR/SAM) | Notes |
|---|---|---|---|
| Typical Gene Knockdown/Efficacy | 70-95% knockdown (mRNA) | 5- to 50-fold activation (mRNA) | Efficacy is highly gene- and context-dependent. |
| On-Target Specificity | High | Moderate | CRISPRa more prone to off-target transcriptional effects. |
| Multiplexing Capability | High (for repression) | Moderate | Simultaneous activation of many genes can be challenging. |
| Inducible Control | Excellent (common) | Good | Often paired with drug-inducible systems (e.g., doxycycline). |
| Kinetics of Effect | Rapid (hours) | Slower (often 24-48 hrs) | Activation requires chromatin remodeling. |
| Therapeutic Applicability (Current) | High (for silencing disease genes) | Emerging (for haploinsufficiency) | CRISPRi is nearer to clinical translation. |
Table 2: Decision Matrix for Tool Selection by Research Objective
| Research Objective | Primary Recommended Tool | Key Rationale | Alternative Consideration |
|---|---|---|---|
| Genome-wide Loss-of-Function Screens | CRISPRi | More consistent, predictable knockdown than RNAi; less toxic than CRISPRn. | CRISPR knockout (CRISPRn) for complete gene ablation. |
| Genome-wide Gain-of-Function Screens | CRISPRa | Enables discovery of genes conferring phenotypes when overexpressed. | cDNA libraries are an alternative but lack genomic context. |
| Modeling Genetic Haploinsufficiency | CRISPRi | Mimics the partial loss of function seen in many disorders. | CRISPRn for complete loss of allele. |
| Modeling Oncogene Activation | CRISPRa | Precise, endogenous gene activation superior to cDNA overexpression. | — |
| Therapy: Silencing Dominant-Negative Alleles | CRISPRi | Safe, reversible suppression of pathogenic gene expression. | CRISPRn for permanent disruption (higher off-target risk). |
| Therapy: Upregulating Protective/Tumor Suppressor Genes | CRISPRa | Endogenous transcriptional activation. | Gene therapy with cDNA transgene. |
Experimental Protocols
Protocol 3.1: Designing and Cloning a Lentiviral sgRNA Library for CRISPRi/a Screens
Objective: To construct a pooled lentiviral sgRNA library for a genome-wide CRISPRi or CRISPRa screen. Materials: See "The Scientist's Toolkit" below. Method:
- sgRNA Design: Use established algorithms (e.g., from the Weissman or Gilbert labs). For CRISPRi, design sgRNAs targeting the transcriptional start site (TSS) -50 to +300 bp relative to the TSS. For CRISPRa, target sgRNAs -400 to -50 bp upstream of the TSS.
- Oligo Pool Synthesis: Order a pooled oligonucleotide library containing the sgRNA sequences, flanked by cloning adapters.
- Library Cloning: Amplify the oligo pool by PCR. Digest the lentiviral backbone plasmid (e.g., lentiGuide-puro for CRISPRi; lentiSAMv2 for CRISPRa) with BsmBI. Purify the linearized vector.
- Golden Gate Assembly: Perform a BsmBI-v2 Golden Gate assembly of the PCR-amplified sgRNA inserts and the digested backbone. Use T7 DNA Ligase and cycle between 37°C and 16°C.
- Transformation & Library Amplification: Transform the assembled reaction into Endura electrocompetent E. coli. Plate on large 245 x 245 mm LB agar plates with appropriate antibiotic. Harvest all colonies to ensure >200x representation of the library.
- Plasmid Purification: Isolate the plasmid library using a maxiprep kit. Validate by next-generation sequencing to confirm sgRNA representation and distribution.
Protocol 3.2: Performing a Pooled Positive Selection Survival Screen
Objective: To identify genes whose knockdown (CRISPRi) or activation (CRISPRa) confer resistance to a drug treatment. Method:
- Virus Production: In a 6-well plate, co-transfect HEK293T cells with the sgRNA library plasmid, psPAX2, and pMD2.G using PEI Max. Harvest lentiviral supernatant at 48 and 72 hours.
- Target Cell Transduction: Plate target cells (e.g., A375 cancer cell line) and transduce with the pooled lentivirus at a low MOI (~0.3) to ensure most cells receive a single sgRNA. Include puromycin selection (e.g., 2 µg/mL) for 5-7 days.
- Selection Phase: Split the selected cell population into two arms: treatment (e.g., with a chemotherapeutic drug at IC90) and control (DMSO). Maintain cells for 14-21 population doublings, ensuring minimum 500x sgRNA representation at all times.
- Genomic DNA Extraction & Sequencing: Harvest ~50 million cells per arm. Extract gDNA using a Blood & Cell Culture DNA Maxi Kit. Perform a two-step PCR to amplify the integrated sgRNA cassettes and add sequencing adapters/indexes.
- Sequencing & Analysis: Sequence on an Illumina NextSeq. Align reads to the sgRNA library reference. Use MAGeCK or similar algorithms to compare sgRNA abundance between treatment and control arms, identifying significantly enriched or depleted sgRNAs.
Visualizations
Decision Matrix Flow for CRISPRa vs CRISPRi
Mechanistic Comparison of CRISPRi and CRISPRa Systems
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPRa/i Research
| Reagent / Material | Function / Description | Example Supplier/Catalog |
|---|---|---|
| dCas9-KRAB Expression Plasmid | Constitutively expresses the CRISPRi repressor fusion protein. | Addgene #71237 (pLV hU6-sgRNA hUbC-dCas9-KRAB-T2A-Puro) |
| dCas9-VPR Expression Plasmid | Constitutively expresses the CRISPRa activator fusion protein. | Addgene #63798 (pHAGE EF1α dCas9-VPR) |
| Lentiviral sgRNA Backbone | Plasmid for cloning sgRNA libraries; contains puromycin resistance. | Addgene #52963 (lentiGuide-Puro for CRISPRi) / #75112 (lentiSAMv2 for CRISPRa) |
| Lentiviral Packaging Plasmids | psPAX2 (gag/pol) and pMD2.G (VSV-G env) for virus production. | Addgene #12260 & #12259 |
| Polybrene (Hexadimethrine bromide) | A cationic polymer that enhances viral transduction efficiency. | Sigma-Aldrich H9268 |
| Puromycin Dihydrochloride | Selective antibiotic for cells transduced with puromycin-resistant vectors. | Thermo Fisher Scientific A1113803 |
| Next-Generation Sequencing Kit | For preparing sgRNA amplicon libraries from genomic DNA (2-step PCR). | Illumina Nextera XT DNA Library Prep Kit |
| MAGeCK Software | Computational tool for analyzing CRISPR screen data (count, test, rank). | https://sourceforge.net/p/mageck/wiki/Home/ |
| Endura Electrocompetent E. coli | High-efficiency bacteria for transforming large, complex plasmid libraries. | Lucigen 60242-2 |
Conclusion
CRISPRa and CRISPRi represent two sides of the same coin, offering researchers unparalleled precision in controlling gene expression without altering the underlying DNA sequence. CRISPRa excels in gain-of-function studies and therapeutic activation of endogenous genes, while CRISPRi provides a potent and reversible alternative to RNAi for knockdowns. Successful implementation requires careful consideration of gRNA design, delivery, and robust validation. As these technologies mature, their integration into multiplexed screens, synthetic biology circuits, and precision medicine initiatives will accelerate. Future directions point toward enhanced specificity, inducible and tissue-specific systems, and clinical translation for diseases requiring precise transcriptional modulation, solidifying their role as indispensable tools in modern biomedical research.