CRISPR-Cas9 vs. Base Editing: A Comparative Guide for Precision Genome Engineering in Research & Therapy
This article provides a comprehensive, comparative analysis of CRISPR-Cas9 and base editing technologies for precision genome engineering, tailored for researchers, scientists, and drug development professionals.
CRISPR-Cas9 vs. Base Editing: A Comparative Guide for Precision Genome Engineering in Research & Therapy
Abstract
This article provides a comprehensive, comparative analysis of CRISPR-Cas9 and base editing technologies for precision genome engineering, tailored for researchers, scientists, and drug development professionals. It explores the foundational molecular mechanisms of each platform, details their methodological workflows and key applications in biomedical research, addresses common challenges and optimization strategies, and conducts a head-to-head validation of their precision, efficiency, and safety profiles. The synthesis aims to empower informed platform selection for specific experimental and therapeutic goals.
Decoding the Molecular Scissors: Core Mechanisms of CRISPR-Cas9 and Base Editing
Within the ongoing thesis comparing CRISPR-Cas9 and base editing for precision genome engineering, understanding the foundational Cas9 paradigm is critical. This guide objectively compares the performance of standard CRISPR-Cas9, which relies on guide RNA (gRNA), creates double-strand breaks (DSBs), and harnesses cellular repair pathways, against alternative precision editing tools, focusing on experimental data relevant to research and therapeutic development.
Comparative Performance: CRISPR-Cas9 vs. Prime Editing and Base Editing
The standard CRISPR-Cas9 system's reliance on DSB repair is both its strength for gene knockout and its limitation for precise point correction. The following table compares key performance metrics with prime editing and adenine base editors (ABEs), two leading alternatives that avoid DSBs.
Table 1: Performance Comparison of CRISPR-Cas9, Base Editing, and Prime Editing
| Metric | CRISPR-Cas9 (NHEJ/HDR) | Adenine Base Editor (ABE8e) | Prime Editor (PE2) |
|---|---|---|---|
| Primary Editing Outcome | Indels (NHEJ) or precise templated edits (HDR) | A•T to G•C conversion | All 12 possible point mutations, small insertions/deletions |
| Double-Strand Break Formation | Yes | No | No |
| Theoretical Editing Precision | Low (NHEJ) / High (HDR) | High | Very High |
| Typical Editing Efficiency Range* (%) | 1-40 (HDR); 10-80 (NHEJ) | 20-80 | 10-50 |
| Indel Byproduct Rate* (%) | 0.5 - 20 (at on-target) | < 1.0 | < 1.0 |
| Product Purity* (%) | Low for HDR | Very High | High |
| Key Limitation | Low HDR efficiency in non-dividing cells; high indel byproducts | Restricted to specific base changes; bystander editing | Large construct; variable efficiency across loci |
*Data compiled from recent primary literature (2022-2024) in mammalian cell lines. Efficiency is locus-dependent.
Experimental Protocols for Key Comparisons
Protocol 1: Measuring On-Target Editing Efficiency and Byproducts
This protocol is used to generate data comparable to Table 1.
- Design & Cloning: Design gRNAs for a target locus. Clone into appropriate Cas9, ABE, or PE expression plasmids.
- Delivery: Transfect target cells (e.g., HEK293T, iPSCs) with editing plasmids and relevant gRNAs/pegRNAs.
- Harvest: Extract genomic DNA 72-96 hours post-transfection.
- Amplification: PCR amplify the target region.
- Analysis: Utilize next-generation sequencing (NGS). For Cas9, assess indel percentage via decomposition tools (e.g., CRISPResso2). For ABE/PE, quantify base conversion percentages. Calculate indel byproduct rates from NGS data for all systems.
Protocol 2: Assessing HDR vs. NHEJ Outcomes in Cas9 Editing
This protocol quantifies the repair pathway choice after Cas9 DSB.
- Setup: Include a donor DNA template for HDR (single-stranded or double-stranded) containing a silent restriction site or a fluorescent reporter.
- Co-delivery: Co-transfect cells with Cas9-gRNA ribonucleoprotein (RNP) and the donor template.
- Analysis: For restriction site assay, perform PCR and restriction digest. For fluorescent reporters, analyze by flow cytometry. The HDR efficiency = (HDR-positive cells / total transfected cells). NHEJ efficiency is derived from indel frequency in the absence of the donor.
Pathway and Workflow Visualizations
Title: CRISPR-Cas9 Double-Strand Break Repair Pathways
Title: Workflow for NGS-Based Editing Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for CRISPR-Cas9 & Comparative Studies
| Reagent / Solution | Function in Experiment | Example Vendor/Product |
|---|---|---|
| High-Fidelity DNA Polymerase | Accurate amplification of target loci for NGS. | NEB Q5, Takara PrimeSTAR GXL |
| Next-Generation Sequencing Kit | Prepare sequencing libraries from PCR amplicons. | Illumina DNA Prep, Swift Biosciences Accel-NGS 2S |
| Cas9 Nuclease (WT) | Generate double-strand breaks for the classic paradigm. | IDT Alt-R S.p. Cas9 Nuclease, Thermo Fisher TrueCut Cas9 Protein v2 |
| Adenine Base Editor (ABE) Plasmid | Enable A•T to G•C editing without DSBs for comparison. | Addgene #138489 (ABE8e), Beam Therapeutics custom mRNAs |
| Prime Editor (PE) Plasmid | Enable versatile point edits & small indels without DSBs. | Addgene #174828 (PE2), Thermo Fisher TrueCut PE2 Protein |
| Chemically Modified gRNA / sgRNA | Enhance stability and editing efficiency. | IDT Alt-R CRISPR-Cas9 sgRNA (2'-O-methyl analogs) |
| Single-Stranded DNA Donor Oligo | Serve as a repair template for HDR experiments. | IDT Ultramer DNA Oligo, Genewiz gBlocks |
| Lipid-based Transfection Reagent | Deliver CRISPR ribonucleoproteins (RNPs) or plasmids to cells. | Thermo Fisher Lipofectamine CRISPRMAX, Mirus Bio TransIT-X2 |
| Genomic DNA Extraction Kit | Cleanly isolate gDNA from transfected cells for analysis. | Qiagen DNeasy Blood & Tissue Kit, Zymo Research Quick-DNA Miniprep Kit |
Thesis Context: CRISPR-Cas9 vs. Base Editing for Precision Genome Engineering
The quest for precision in genome engineering has evolved from the double-strand break (DSB)-dependent CRISPR-Cas9 system to DSB-free base editing technologies. While CRISPR-Cas9 facilitates gene knockouts via non-homologous end joining (NHEJ), its reliance on DSBs leads to unintended indels and makes precise single-nucleotide corrections inefficient. Base editors (BEs), constructed by fusing a catalytically impaired Cas9 (dCas9 or nickase Cas9) to a deaminase enzyme, directly convert one DNA base pair to another without creating DSBs, offering superior precision for single-nucleotide variant (SNV) correction.
Core Mechanism and Comparison to Alternatives
Base editors are classified primarily into Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs). CBEs use a cytidine deaminase to convert C•G to T•A, while ABEs use an engineered adenine deaminase to convert A•T to G•C. This section compares their performance against conventional CRISPR-Cas9 homology-directed repair (HDR) and prime editing.
Table 1: Performance Comparison of Major Genome Engineering Tools
| Feature | CRISPR-Cas9 HDR | Cytosine Base Editor (CBE) | Adenine Base Editor (ABE) | Prime Editor (PE) |
|---|---|---|---|---|
| Primary Edit Type | All possible changes | C•G to T•A | A•T to G•C | All 12 possible base-to-base conversions, small insertions/deletions |
| Requires DSB? | Yes | No | No | No |
| Requires Donor Template? | Yes | No | No | Yes (pegRNA) |
| Typical Efficiency in Mammalian Cells (%) | 0.1–20% (low) | 15–75% (high) | 15–50% (high) | 10–50% (moderate) |
| Indel Formation (%) | High (often >10%) | Low (<1% for latest gens) | Very Low (<1%) | Very Low (<1%) |
| Product Purity | Low | High | Very High | High |
| Common Off-Targets | DNA DSB sites, gRNA-dependent | gRNA-independent RNA off-targets (CBE v1), gRNA-dependent DNA | Minimal RNA off-targets | gRNA-dependent DNA |
| Key Limitation | Low efficiency, requires cell cycle, donor delivery | Restricted to C•G to T•A edits, potential C-to-T bystander edits within window | Restricted to A•T to G•C edits | Larger construct, more complex gRNA design |
Table 2: Experimental Data from Key Studies (Representative)
| Study (Year) | Editor Tested | Target/Gene | Cell Type | Editing Efficiency (%) | Indel Rate (%) | Purity (Desired Product/Total Edited) |
|---|---|---|---|---|---|---|
| Komor et al. (2016) | BE3 (CBE) | HEK293 site 4 | HEK293T | 37% | ~1.3 | ~67% |
| Gaudelli et al. (2017) | ABE7.10 | HEK293 site 4 | HEK293T | 53% | <0.1 | >99.9% |
| Anzalone et al. (2019) | PE2 | HEK293 site 3 | HEK293T | 20–50% | <0.1 | 78% |
| Grunewald et al. (2019) | BE4max (CBE) | VEGFA site | HEK293T | 74% | 0.05 | ~85% |
| Newby et al. (2021) | ABE8e | PCSK9 | Primary Hepatocytes | 65% | 0.04 | >99.9% |
Detailed Experimental Protocols
Protocol 1: Evaluating CBE Editing Efficiency and Byproduct Formation Objective: Quantify targeted C-to-T conversion efficiency and indel byproduct formation at an endogenous locus. Materials: HEK293T cells, BE4max plasmid, targeting gRNA plasmid, transfection reagent, genomic DNA extraction kit, PCR primers flanking target site, T7 Endonuclease I (T7EI) or Surveyor nuclease, agarose gel, Sanger sequencing reagents, next-generation sequencing (NGS) library prep kit. Method:
- Transfection: Co-transfect HEK293T cells with BE4max and target-specific gRNA plasmids using a cationic polymer.
- Harvest: 72 hours post-transfection, harvest cells and extract genomic DNA.
- PCR Amplification: Amplify the target genomic region with high-fidelity polymerase.
- Analysis:
- T7EI Assay: Hybridize PCR products, digest with T7EI, analyze fragments on agarose gel to estimate total editing (indels + base edits).
- NGS Analysis: Barcode and amplify PCR products for Illumina sequencing. Analyze sequencing reads for precise C-to-T conversions, bystander C edits within the editing window (typically ~5 nucleotides), and indel percentages.
- Calculation: Efficiency = (Number of reads with target C-to-T) / (Total reads) * 100%. Purity = (Target C-to-T reads) / (All edited reads including bystanders and indels) * 100%.
Protocol 2: Assessing RNA Off-Targets in Base Editors Objective: Identify transcriptome-wide RNA cytosine deamination caused by CBEs. Materials: Cells transfected with CBE (e.g., BE3) or ABE control, total RNA extraction kit, cDNA synthesis kit, PCR reagents for known off-target sites, or materials for whole-transcriptome RNA sequencing (RNA-seq). Method:
- Treatment: Prepare two sets of cells: one transfected with CBE+gRNA, another with ABE+gRNA as a deaminase-activity control.
- RNA Sequencing: 48 hours post-transfection, extract total RNA. Perform poly-A selection, library preparation, and whole-transcriptome sequencing.
- Bioinformatics Analysis: Map RNA-seq reads to the transcriptome. Use variant callers to identify C-to-U changes. Filter out known SNPs and changes present in the ABE control sample.
- Validation: For candidate off-target RNA sites, perform targeted cDNA amplification and deep sequencing.
Visualization of Base Editor Mechanism and Workflow
Diagram 1: CBE Mechanism (76 chars)
Diagram 2: BE Experimental Workflow (64 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Base Editing Research
| Item | Function/Application | Example Vendor/Product |
|---|---|---|
| Base Editor Plasmids | Provide the gene encoding the dCas9-deaminase fusion protein for mammalian expression. | Addgene (pCMVBE4max, pCMVABE8e) |
| gRNA Cloning Vector | Backbone for expressing the target-specific single guide RNA (sgRNA). | Addgene (pU6-sgRNA, pX330-derived) |
| Delivery Reagent | Transfect plasmid or RNP into hard-to-transfect cells (e.g., primary cells). | Lipofectamine CRISPRMAX, Lonza Nucleofector |
| Purified Base Editor Protein | For forming Ribonucleoprotein (RNP) complexes for delivery with reduced off-target persistence. | Aldevron, Thermo Fisher TrueCut Cas9 Protein v2 + chemical conjugation to deaminase |
| NGS-based Editing Analysis Service | Quantifies editing efficiency, bystander edits, and indels with high accuracy. | IDT xGen NGS, Genewiz Amplicon-EZ |
| Control gRNA & Target Plasmids | Validated positive and negative controls for assay optimization. | Synthego EZ Edit Control Kit |
| Genomic DNA Extraction Kit | High-yield, PCR-ready DNA from cultured cells. | Qiagen DNeasy Blood & Tissue Kit |
| High-Fidelity PCR Master Mix | Accurate amplification of target locus for sequencing analysis. | NEB Q5, KAPA HiFi HotStart |
| T7 Endonuclease I | Fast, gel-based assay to estimate total editing activity at a locus. | NEB T7 Endonuclease I |
| RNA Deaminase Inhibitor | Chemical to suppress RNA off-target activity of certain CBEs in sensitive applications. | rAPOBEC1 inhibitor (Research compounds) |
Within the ongoing debate on CRISPR-Cas9 versus base editing for precision genome engineering, understanding the protein architecture that enables each technology is fundamental. This guide compares the structural and functional evolution from Cas9 nickases to engineered deaminases, highlighting the performance implications.
Core Protein Architectures and Performance Data
| Feature | CRISPR-Cas9 Nickase (e.g., D10A or H840A) | DNA Base Editor (e.g., BE4) | RNA Base Editor (e.g., REPAIRv2) |
|---|---|---|---|
| Core Catalytic Domain | RuvC or HNH (one inactive) | Deoxycytidine Deaminase (e.g., rAPOBEC1) | Adenosine Deaminase (e.g., ADAR2 dd) |
| Targeting Module | Cas9 domain + gRNA | Cas9 nickase domain + gRNA | dCas13 (or inactivated Cas13) + gRNA |
| Key Accessory Domains | None (minimal) | UGI (Uracil Glycosylase Inhibitor): Prevents base excision repair. | Linker & Localization Sequences: Optimizes efficiency. |
| Primary Function | Creates single-strand DNA break (nick) | Converts C•G to T•A (CBE) or A•T to G•C (ABE)* | Converts A to I (read as G) in RNA |
| Edit Type | Triggers HDR or bias MMR | Permanent DNA point mutation without DSB | Transient RNA alteration; no genomic change |
| Typical Editing Efficiency (from cited studies) | Low HDR (<10%) | High (30-70%) for CBE/ABE in mammalian cells | High (20-50%) for transcript editing |
| Indel Byproduct | Moderate (from residual DSBs) | Very Low (<1%) with optimized editors | None (RNA is not replicated) |
| PAM/Restriction | SpCas9 PAM (NGG) required | SpCas9 PAM (NGG) required; broader PAM variants available | Minimal PAM preference (proximal to editable base) |
*ABEs use an evolved TadA adenosine deaminase.
Experimental Protocols for Key Validation Studies
1. Protocol: Measuring DNA Base Editing Efficiency and Product Purity
- Objective: Quantify target base conversion frequency and indel byproduct formation.
- Method:
- Transfection: Deliver base editor plasmid (e.g., BE4) and target-specific gRNA into HEK293T cells.
- Harvest: Extract genomic DNA 72 hours post-transfection.
- PCR Amplification: Amplify the target genomic locus.
- Sequencing: Perform Sanger or next-generation sequencing (NGS) of the PCR amplicon.
- Analysis: Use computational tools (e.g., BE-Analyzer, CRISPResso2) to calculate the percentage of C-to-T (or A-to-G) conversion and the percentage of indel-containing reads within the treated sample.
2. Protocol: Assessing RNA Base Editing Specificity
- Objective: Determine transcriptome-wide off-target editing by an RNA base editor.
- Method:
- Treatment: Express the RNA editor (e.g., REPAIRv2) in relevant cell lines.
- RNA-Seq: 48 hours later, perform total RNA sequencing with high depth.
- Bioinformatics: Align sequences to the reference genome/transcriptome. Use variant calling pipelines (e.g., GATK) specifically tuned to identify A-to-G mismatches, which indicate A-to-I editing.
- Filtering: Filter for A-to-G changes in the absence of DNA variants, then compare against a negative control sample to identify editor-dependent events.
Visualizations
The Scientist's Toolkit: Key Research Reagents
| Reagent/Material | Function in Experiment |
|---|---|
| Base Editor Plasmid (e.g., BE4, ABE8e) | Expresses the fusion protein containing nickase, deaminase, and accessory domains. |
| sgRNA Expression Construct | Encodes the guide RNA for specific target site localization. |
| Delivery Vehicle (e.g., PEI, Lipofectamine, Electroporator) | Facilitates intracellular delivery of editor machinery into cultured cells. |
| Control Plasmids (Wild-type Cas9, Nickase only) | Essential controls to compare efficiency and specificity against base editors. |
| NGS Library Prep Kit | Prepares amplified target DNA for high-throughput sequencing to quantify editing. |
| Variant Analysis Software (CRISPResso2, BE-Analyzer) | Specialized bioinformatics tools to deconvolve complex editing outcomes from sequencing data. |
| Cell Line with Stably Integrated Reporter | Provides a rapid, fluorescence-based preliminary assessment of editing efficiency. |
This guide provides an objective comparison of two dominant precision genome engineering technologies: CRISPR-Cas9-mediated homology-directed repair (HDR) and DNA base editing. The core trade-off lies between the versatile, DSB-dependent pathway of traditional CRISPR-Cas9 and the DSB-free, chemistry-driven point mutation approach of base editors.
Core Technology Comparison
Table 1: Fundamental Characteristics and Performance Metrics
| Feature | CRISPR-Cas9 HDR Editing | Adenine Base Editor (ABE) | Cytosine Base Editor (CBE) |
|---|---|---|---|
| Editing Chemistry | Cellular HDR machinery | tRNA adenosine deaminase | APOBEC cytidine deaminase |
| Double-Strand Break (DSB) | Required | Not required | Not required |
| Primary Edit Type | Targeted insertions, deletions, substitutions (all 12 possible) | A•T to G•C transition | C•G to T•A transition |
| Typical Efficiency (in cultured mammalian cells) | 1-20% (highly variable) | 20-50% (can exceed 75%) | 15-40% (context-dependent) |
| Indel Byproduct Formation | High (>5-20%) | Very low (<1%) | Low to moderate (1-10%) |
| Product Purity | Low (mixed outcomes common) | Very High | High |
| Theoretical Targetable Positions in Human Genome* | ~100% | ~25% (requires an A in the editing window) | ~15% (requires a C in the editing window) |
| Key Limitation | Low efficiency, high indel rate, active cell cycle required | Restricted to A-to-G edits | Restricted to C-to-T edits; potential C•G to G•C, C•G to A•T transversions |
*Based on the presence of a suitable PAM sequence (e.g., NGG for SpCas9) and a protospacer.
Experimental Data from Key Studies
Table 2: Comparative Experimental Data from Recent Studies
| Parameter | Study (Cell Type) | CRISPR-Cas9 HDR Result | Base Editor (BE) Result | Key Takeaway |
|---|---|---|---|---|
| Point Correction Efficiency | Gaudelli et al., 2017 (HEK293T) | HDR: ~1.7% correction, >10% indels | ABE7.10: ~50% correction, <0.1% indels | ABE achieved 28x higher correction with minimal indels. |
| Editing in Non-Dividing Cells | Koblan et al., 2021 (Post-mitotic mouse neurons) | HDR: Negligible correction | AAV-delivered ABE: ~35% correction in vivo | Base editors function effectively in non-dividing cells. |
| Off-target DNA Editing | Zuo et al., 2019 (Whole-genome sequencing) | Cas9: DSB-dependent indels at related genomic sites | BE3: No significant increase in sgRNA-dependent mutations | Catalytically impaired Cas9 in BEs reduces off-target DNA cleavage. |
| On-target Product Purity | Komor et al., 2016 (HEK293T) | HDR for C-to-T: Low, with high indel background | BE3: >99% C-to-T products within edited population | BEs offer precise single-base changes without DSB-related byproducts. |
| Therapeutic in vivo Editing | Villiger et al., 2018 (Mouse liver, PCSK9 KO) | SaCas9 HDR: ~2% gene correction, ~30% indels | SaBE: ~60% gene correction, <1% indels | BE delivery yielded superior correction rates and cleaner outcomes. |
Detailed Experimental Protocols
Protocol 1: CRISPR-Cas9 HDR for Point Mutation Installation
This protocol is for introducing a specific point mutation using a single-stranded oligodeoxynucleotide (ssODN) donor.
- Design Components: Design a sgRNA targeting the genomic locus immediately adjacent to the desired mutation. Design a homology-directed repair template (ssODN, ~100-200 nt) encoding the desired point mutation, flanked by ~40-80 nt homology arms on each side. Incorporate silent mutations in the PAM/protospacer to prevent re-cutting.
- Cell Transfection: Seed HEK293T cells in a 24-well plate. At 70-80% confluency, co-transfect using a suitable reagent (e.g., Lipofectamine 3000):
- 500 ng Cas9 expression plasmid or 250 ng Cas9 RNP (ribonucleoprotein)
- 250 ng sgRNA expression plasmid or 100 pmol synthetic sgRNA (if using RNP)
- 100 pmol of Ultramer ssODN donor template.
- Harvest and Analyze: Harvest cells 48-72 hours post-transfection. Extract genomic DNA. Analyze editing efficiency via next-generation sequencing (NGS) of PCR-amplified target loci. Calculate HDR efficiency as (# of reads with precise edit) / (total aligned reads). Quantify indel frequency using tools like CRISPResso2.
Protocol 2: Base Editing for Targeted Transition Mutation
This protocol is for installing an A-to-G or C-to-T mutation using a plasmid-based base editor.
- Design Components: Design a sgRNA positioning the target base within the effective editing window (typically positions 4-8 for SpCas9-derived BEs, counting from the PAM-distal end). No donor template is required.
- Cell Transfection: Seed HEK293T cells in a 24-well plate. At 70-80% confluency, transfect using a suitable reagent:
- 500 ng base editor expression plasmid (e.g., ABE8e or BE4max)
- 250 ng sgRNA expression plasmid.
- Harvest and Analyze: Harvest cells 48-72 hours post-transfection. Extract genomic DNA. Amplify the target region by PCR and submit for NGS. Calculate base editing efficiency as (# of reads with desired transition) / (total aligned reads). Analyze for bystander edits (other bases changed within the window) and low-frequency indels.
Visualizing the Core Trade-off and Mechanisms
Title: CRISPR-Cas9 HDR Pathway Leads to Mixed Outcomes
Title: Base Editing Uses Chemical Conversion for Precision
Title: Core Trade-off Between Editing Platforms
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Comparative Studies
| Reagent | Function in CRISPR-Cas9 HDR | Function in Base Editing | Example Vendor/Product |
|---|---|---|---|
| Nuclease | Wild-type SpCas9: Creates a DSB at the target site. | Nickase Cas9 (nCas9, D10A): Creates a single-strand break for base editors like BE4. Catalytically dead Cas9 (dCas9): No cleavage; used for targeting only (e.g., in ABE8e). | IDT, Thermo Fisher, Addgene plasmids. |
| Deaminase Enzyme | Not used. | Cytidine Deaminase (e.g., rAPOBEC1): Converts C to U in CBEs. Adenosine Deaminase (e.g., TadA): Converts A to I in ABEs. | Encoded within base editor plasmids from Addgene. |
| sgRNA | Guides Cas9 to the target genomic locus. | Guides the base editor complex to the target locus, positioning the editing window. | Synthesized as crRNA:tracrRNA duplex or as a single guide (sgRNA) from IDT, Synthego. |
| Repair Template | ssODN or dsDNA donor: Provides the homologous sequence for HDR to copy the desired edit. | Not required. The chemical conversion is encoded by the editor complex itself. | Ultramer ssODNs from IDT; dsDNA fragments. |
| Delivery Vehicle | Plasmids, RNPs, or viral vectors (lentivirus, AAV) for in vitro/vivo delivery. | Plasmids, RNPs, or viral vectors (AAV preferred for in vivo due to smaller size constraints of some BEs). | Lipofectamine (plasmid), JetMessenger (RNP), AAVpro (viral). |
| Analysis Tool | NGS + CRISPResso2: Quantifies HDR efficiency and indel spectrum from DSB repair. | NGS + BE-Analyzer/BEAT: Quantifies base conversion efficiency and bystander editing profiles. | Open-source software tools. |
From Bench to Bedside: Protocols and Cutting-Edge Applications of Each Technology
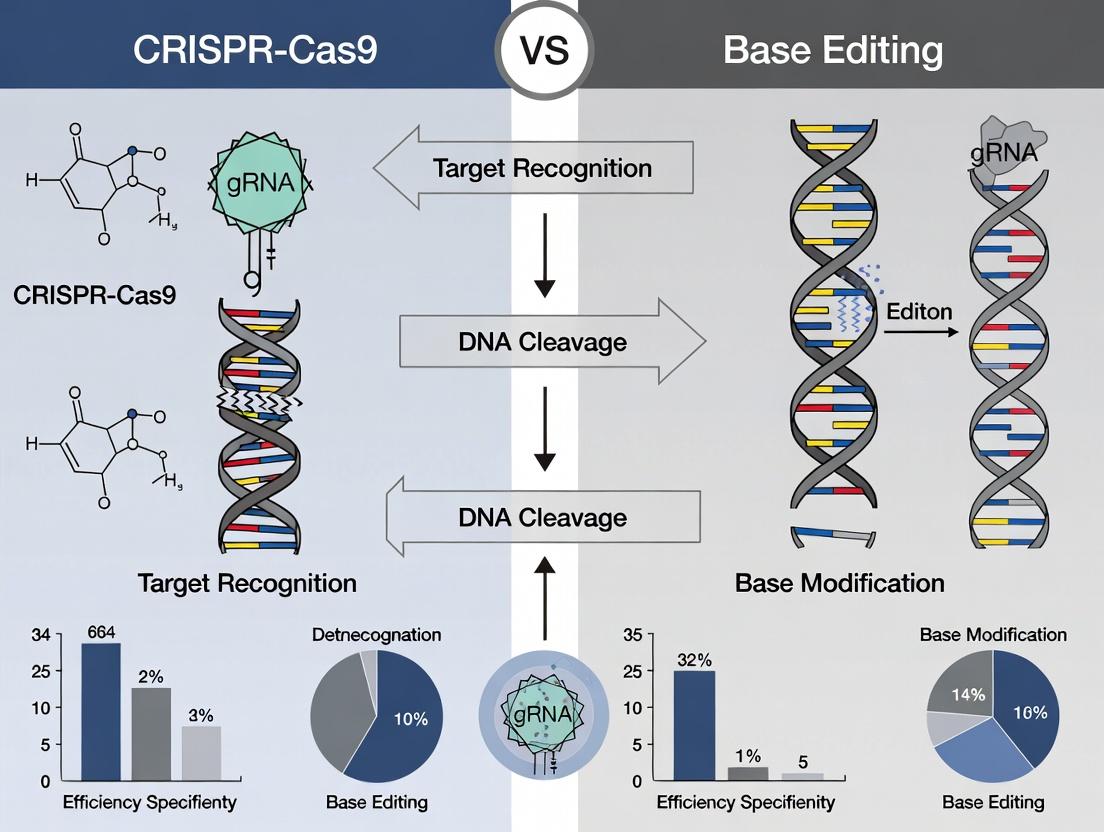
The choice of genome editing technology is critical for precision research. While base editing offers direct chemical conversion of nucleotides without double-strand breaks (DSBs), the canonical CRISPR-Cas9 system remains the most widely adopted for gene knockout and knock-in studies. This guide compares the standard CRISPR-Cas9 workflow against alternative methods, framing performance within the broader thesis of CRISPR-Cas9 versus base editing for precision outcomes.
Workflow Comparison: CRISPR-Cas9 vs. Base Editing
The fundamental divergence occurs after target recognition. The standard CRISPR-Cas9 workflow is predicated on generating a DSB, leading to repair outcomes that can be heterogeneous. Base editing bypasses the DSB, directly converting one base pair to another.
Diagram Title: CRISPR-Cas9 vs Base Editing Workflow Divergence
Performance Comparison: Editing Efficiency and Outcome Purity
Recent head-to-head studies for correcting point mutations illustrate key trade-offs.
Table 1: Comparison of Editing Outcomes at the EMXI Locus (HEK293T Cells)
| Parameter | CRISPR-Cas9 (HDR with ssODN) | Adenine Base Editor (ABE8e) | Cytosine Base Editor (BE4max) |
|---|---|---|---|
| Target Modification | A•T to G•C | A•T to G•C | C•G to T•A |
| Average Editing Efficiency | 12.5% ± 3.2% | 58.7% ± 5.1% | 44.3% ± 4.8% |
| Precise Desired Product | 8.1% ± 2.7% | 55.9% ± 4.9% | 41.0% ± 4.5% |
| Indel Byproducts | 31.0% ± 6.5% | <1.0% | 1.2% ± 0.4% |
| Bystander Edits | N/A | Low (within window) | Moderate (within window) |
Experimental Protocol for Table 1 Data:
- Design: sgRNAs were designed adjacent to the target nucleotide. For Cas9-HDR, a 120-nt ssODN donor with homology arms and the desired base change was synthesized.
- Delivery: HEK293T cells were co-transfected via lipofection with: a) Cas9 plasmid + sgRNA plasmid + ssODN, or b) Base editor plasmid (ABE8e or BE4max) + sgRNA plasmid.
- Screening: 72 hours post-transfection, genomic DNA was harvested. The target locus was PCR-amplified and analyzed by next-generation sequencing (NGS).
- Validation: NGS reads were analyzed for precise base conversion, total editing (all sequence changes), and indel frequency. Data from three biological replicates were pooled.
Table 2: Comparative Analysis for Functional Knockouts
| Parameter | CRISPR-Cas9 (NHEJ) | CRISPR-Cas9 (HDR - STOP cassette) | Base Editing (Introducing Premature STOP) |
|---|---|---|---|
| Knockout Efficiency | High (70-95%) | Moderate (10-30%) | Variable (5-50%)* |
| Clonal Homogeneity | Low (Mixed Indels) | High (Precise Insertion) | High (Precise Point Mutation) |
| Multiplexing Ease | High | Moderate | High |
| Off-target Genomic Risk | DSB-dependent & DSB-independent | DSB-dependent & DSB-independent | DSB-independent only |
*Efficiency depends on presence of a convertible codon within the editable window.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for the Standard CRISPR-Cas9 Workflow
| Reagent | Function | Key Considerations for Comparison |
|---|---|---|
| SpCas9 Nuclease | Creates the double-strand break at the target site. | Wild-type vs. high-fidelity variants (e.g., SpCas9-HF1) to balance on-target efficiency and off-target reduction. |
| sgRNA (synthetic or expressed) | Guides Cas9 to the specific genomic locus. | Chemical modification (e.g., 2'-O-methyl analogs) can enhance stability and efficiency, especially for RNP delivery. |
| HDR Donor Template | Provides the template for precise repair. | ssODN vs. double-stranded DNA donors; length and homology arm optimization are critical for efficiency. |
| Delivery Vehicle | Introduces editing components into cells. | Lipofection reagents, electroporation systems (e.g., Neon), or viral vectors (AAV, lentivirus) chosen based on cell type. |
| Enrichment & Screening Tools | Isolates and identifies edited cells. | Fluorescent reporters (e.g., GFP dropout), antibiotic resistance markers, or surface epitope tags for FACS/MACS. |
| Validation Assays | Confirms on-target edit and checks for off-target effects. | NGS-based amplicon sequencing (for on-target), GUIDE-seq or Digenome-seq (for unbiased off-target profiling). |
Diagram Title: CRISPR-Cas9 Screening and Validation Cascade
The standard CRISPR-Cas9 workflow excels at generating complete gene knockouts via NHEJ and enables flexible knock-ins via HDR, albeit with variable efficiency and indel byproducts. Base editing provides superior efficiency and purity for precise point mutations without DSBs but is constrained by its editing window and compatible base changes. The choice hinges on the research goal: for scalable knockouts, Cas9-NHEJ is robust; for point mutation correction, base editors are often superior; for precise sequence insertions, Cas9-HDR remains necessary. An integrated validation cascade is mandatory for both to ensure on-target fidelity.
This guide provides a comparative analysis of base editing protocols within the broader thesis of CRISPR-Cas9 versus base editing for precision genome engineering. We focus on three critical performance parameters: the effective targeting window, editing efficiency, and the purity of the desired product (i.e., minimization of indels and bystander edits).
Comparison of Base Editor Performance Metrics
The following table summarizes quantitative data from recent head-to-head studies comparing common cytosine base editors (CBEs) and adenine base editors (ABEs), alongside standard CRISPR-Cas9 homology-directed repair (HDR) for point mutations.
Table 1: Performance Comparison of Base Editors and CRISPR-Cas9 HDR
| System (Example) | Primary Edit | Typical Targeting Window (from PAM) | Average Efficiency (Range) | Desired Product Purity (Indels %) | Bystander Edit Frequency |
|---|---|---|---|---|---|
| CRISPR-Cas9 + HDR | Any point mutation | N/A (site-specific) | 1-20% (highly variable) | Often <10% | N/A |
| BE4max (CBE) | C•G to T•A | Protospacer positions 4-10 (CBE) | 30-70% | 95-99% (Indels: ~1%) | Moderate to High in window |
| ABEmax (ABE) | A•T to G•C | Protospacer positions 4-9 (ABE) | 40-80% | >99% (Indels: <0.5%) | Low to Moderate |
| evoFERNY (CBE) | C•G to T•A | Protospacer positions 4-10 | 50-75% | >99% (Indels: <1%) | Reduced |
| SaKKH-BE3 (CBE) | C•G to T•A | Expands to include GC context | 20-50% | ~95% (Indels: ~5%) | Moderate |
Key Insight: Base editors consistently offer higher efficiency and product purity for their specific conversions compared to Cas9-HDR, but are constrained by a narrower, protocol-defined targeting window and can suffer from bystander edits within that window.
Experimental Protocols for Key Analyses
Protocol 1: Determining Editing Efficiency and Purity by Next-Generation Sequencing (NGS)
Objective: Quantify base editing percentage, indel frequency, and bystander edits at the target locus.
- Sample Preparation: 72 hours post-transfection of base editor reagents into cultured cells, harvest genomic DNA.
- PCR Amplification: Design primers to amplify a ~300-400 bp region surrounding the target site. Use high-fidelity polymerase.
- NGS Library Prep: Barcode amplicons from different samples/sites. Purify and pool libraries.
- Sequencing: Perform paired-end sequencing on an Illumina platform to achieve >50,000x read depth per sample.
- Data Analysis: Align reads to the reference genome. Use analysis pipelines (e.g., CRISPResso2, BE-Analyzer) to calculate:
- % Edited Reads: Reads containing target C-to-T or A-to-G conversion.
- % Indel Reads: Reads with insertions/deletions at the target site.
- % Bystander Edits: Reads with additional base edits within the editing window.
- Product Purity: (% Target Edit Reads) / (% Target Edit Reads + % Indel Reads) * 100.
Protocol 2: In Vitro Determination of Targeting Window & Bystander Activity
Objective: Map the precise boundaries of deamination activity for a base editor variant.
- Oligo Substrate Design: Synthesize a double-stranded DNA oligo containing the target protospacer sequence with a randomized 6-10 nucleotide region within the putative editing window.
- In Vitro Editing Reaction: Incubate the oligo substrate with purified base editor protein (e.g., BE4max, ABEmax) and necessary buffers.
- Sequencing & Analysis: Harvest DNA, amplify the randomized region, and perform high-depth NGS. The editing efficiency at each position (for the target base) reveals the enzymatic targeting window and inherent propensity for bystander edits.
Visualization of Base Editing Workflow and Key Concepts
Title: Base Editing Experimental Workflow and Outcomes
Title: Base Editor Targeting Window Relative to PAM
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Base Editing Analysis
| Item | Function/Description | Example Vendor/Product |
|---|---|---|
| High-Fidelity DNA Polymerase | For error-free amplification of target loci for NGS. Critical for accurate background measurement. | NEB Q5, Takara PrimeSTAR GXL |
| CRISPR-Cas9 & Base Editor Plasmids | Delivery vectors for editor expression (e.g., BE4max, ABEmax). | Addgene (non-profit repository) |
| Synthetic sgRNAs | Chemically modified for stability; defines target specificity. | Synthego, IDT, Horizon Discovery |
| RNP Complex Components | Purified Cas9n/base editor protein and sgRNA for ribonucleoprotein delivery. | IDT Alt-R S.p. HiFi Cas9 Nuclease, ToolGen proteins |
| NGS Library Prep Kit | For preparing amplicon libraries from genomic DNA. | Illumina TruSeq, Swift Biosciences Accel-NGS |
| CRISPR Analysis Software | Computational tools to quantify editing outcomes from NGS data. | CRISPResso2, BE-Analyzer, ICE (Synthego) |
| Cell Line Engineering Service | For generating stable, clonal edited cell lines for downstream assays. | Takara Bio, Charles River Labs |
Within the ongoing evaluation of CRISPR-Cas9 versus base editing for precision genome engineering, a key distinction lies in their fundamental approaches. Base editing enables direct, single-nucleotide conversion without requiring double-strand breaks (DSBs). In contrast, classical CRISPR-Cas9 relies on the creation of a targeted DSB, which is then resolved by cellular repair pathways to generate a spectrum of edits. This guide objectively compares the outcomes, efficiencies, and experimental parameters of three primary applications stemming from the DSB-repair paradigm: gene knockouts, large deletions, and precise knock-ins via Homology-Directed Repair (HDR).
Performance Comparison: Key Metrics and Experimental Data
Table 1: Comparison of CRISPR-Cas9 Application Outcomes
| Application | Primary Repair Pathway | Typical Edit Size | Average Efficiency Range (Mammalian Cells) | Key Outcome | Major Byproduct/Challenge |
|---|---|---|---|---|---|
| Gene Knockout | Non-Homologous End Joining (NHEJ) | 1bp - 50bp indels | 40-80% (varies by locus/cell type) | Frameshift mutations, premature stop codons | Incomplete knockout (mixed population) |
| Large Deletion | Microhomology-Mediated End Joining (MMEJ) or NHEJ | 100bp - 1Mb+ | 10-50% (decreases with size) | Removal of regulatory elements, exons, or entire genes | Complex rearrangements, inversions |
| Knock-in via HDR | Homology-Directed Repair (HDR) | Precise insertion (single bp to >1kb) | 1-20% (lower in non-dividing cells) | Precise sequence integration (tags, reporters, corrections) | Predominant NHEJ at the DSB site |
Table 2: Experimental Parameters from Recent Studies (2023-2024)
| Study Focus | Cell Line | Cas9 Delivery | gRNA Design | HDR Template Design | Reported Efficiency | Key Optimization |
|---|---|---|---|---|---|---|
| Knockout (PD-1) | Primary human T cells | RNP electroporation | 2 gRNAs targeting exon 1 | N/A | 75% KO (flow cytometry) | High-fidelity Cas9 variant reduced off-targets by 50-fold. |
| Large Deletion (200kb) | HEK293T | Plasmid transfection | Dual gRNAs spaced 200kb apart | N/A | 22% deletion (PCR assay) | Synchronized Cas9 expression from a single plasmid. |
| Knock-in (GFP tag) | iPSCs | mRNA + ssODN | Single gRNA near stop codon | 100bp ssODN with homology arms | 15% HDR (NGS) | Cell cycle synchronization at S/G2 phase doubled HDR rate. |
| Knock-in (Disease Correction) | Patient-derived fibroblasts | AAVS1-saCas9 + AAV6 donor | Dual gRNAs for DSB & AAVS1 safe harbor | AAV6 vector with 1kb homology arms | 8% targeted integration (qPCR) | Inhibition of NHEJ with small molecule (SCR7) increased HDR by 3x. |
Experimental Protocols
Protocol 1: Generating Gene Knockouts via NHEJ
- Design: Select a target sequence within an early coding exon using validated design tools (e.g., CRISPick). Prioritize on-target score and minimize off-target predictions.
- Complex Formation: Combine 10µg of purified SpCas9 protein with a 1.2x molar ratio of synthetic sgRNA. Incubate at 25°C for 10 minutes to form the Ribonucleoprotein (RNP) complex.
- Delivery: Electroporate the RNP complex into 1x10⁶ target cells using cell-type-specific electroporation parameters.
- Analysis: After 72 hours, extract genomic DNA. Assess editing efficiency via T7 Endonuclease I (T7E1) assay or Tracking of Indels by Decomposition (TIDE) analysis of PCR-amplified target locus. For clonal analysis, single-cell sort and expand colonies for Sanger sequencing.
Protocol 2: Creating Large Deletions with Dual gRNAs
- Design: Design two gRNAs targeting the boundaries of the genomic region to be deleted. Verify individual gRNA activity prior to pairing.
- Cloning: Clone expression cassettes for both gRNAs into a single plasmid backbone expressing SpCas9, or prepare two separate RNP complexes.
- Transfection: Co-deliver both gRNA constructs (or RNPs) into the target cells at equimolar ratios.
- Validation: After 5-7 days, perform genomic PCR with primers flanking the outside of the deletion boundaries. A successful deletion yields a smaller PCR product. Confirm the junction sequence by Sanger sequencing.
Protocol 3: Precise Knock-in via HDR using ssODN Donor
- Design: Design a single gRNA to cut as close as possible to the intended edit site. Synthesize a single-stranded oligodeoxynucleotide (ssODN) donor template (90-200nt) containing the desired edit flanked by homology arms (35-50nt each).
- Cell Cycle Synchronization: Treat actively dividing cells with 2mM thymidine or a CDK1 inhibitor to enrich for cells in S/G2 phase, where HDR is active.
- Co-delivery: Electroporate cells with the Cas9 RNP complex and a 10:1 molar excess of the ssODN donor template.
- NHEJ Inhibition: Add 5µM SCR7 or 1µM NU7026 to the culture media for 24-48 hours post-electroporation to suppress NHEJ.
- Screening: After expansion, screen clones by PCR and sequence analysis for precise integration. For fluorescent reporters, analyze by flow cytometry 5-7 days post-delivery.
Visualizations
Title: Gene Knockout via NHEJ Workflow
Title: DSB Repair Pathways for CRISPR Applications
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CRISPR-Cas9 Editing Applications
| Reagent / Solution | Function / Purpose | Example Product/Format |
|---|---|---|
| High-Efficiency Cas9 Nuclease | Generates the targeted double-strand break. Critical for all applications. | Purified S. pyogenes Cas9 protein (RNP ready), HiFi Cas9 variants. |
| Chemically Modified sgRNA | Increases stability and reduces immunogenicity in cells, improving RNP efficiency. | Synthetic sgRNA with 2'-O-methyl 3' phosphorothioate modifications. |
| HDR Donor Templates | Provides the homologous sequence for precise repair. Format depends on edit size. | Ultramer ssODNs (<200nt), dsDNA fragments (PCR/gBlock), AAV or plasmid donors. |
| NHEJ Inhibitors | Small molecules that temporarily inhibit the NHEJ pathway to favor HDR. | SCR7, NU7026, RS-1. Typically used for knock-in experiments. |
| Electroporation Enhancer | Improves viability and delivery efficiency in hard-to-transfect cells. | Commercial electroporation supplements (e.g., CloneOne). |
| Editing Validation Assay | Quantifies indels and HDR efficiency at the target locus. | T7E1 assay kit, TIDE analysis software, NGS-based amplicon sequencing service. |
| Clonal Isolation Medium | Supports the growth and expansion of single-cell clones after editing. | Conditioned media or commercial clone recovery supplements. |
Within the ongoing thesis comparing CRISPR-Cas9 and base editing for precision genome engineering, this guide objectively compares the performance of leading base editor platforms. Base editing enables direct, irreversible conversion of one DNA base pair to another without requiring double-stranded DNA breaks (DSBs), minimizing indel formation. This is critical for applications requiring high-fidelity single-nucleotide variant (SNV) installation or correction.
Performance Comparison of Major Base Editor Systems
The following tables summarize key performance metrics from recent head-to-head studies for cytosine base editors (CBEs) and adenine base editors (ABEs).
Table 1: Comparison of Cytosine Base Editors (C→G, C→T)
| Base Editor System | Deaminase Domain | Average Editing Efficiency (%) (Reported Range) | Product Purity (Desired C→T vs. Indels) | Primary Byproducts | Key Reference (Year) |
|---|---|---|---|---|---|
| BE4max | rAPOBEC1 | 50-80% | High (>99:1) | C→G, C→A | Koblan et al., 2021 |
| Target-AID | pmCDA1 | 20-50% | Moderate | C→G, C→A | Nishida et al., 2016 |
| evoFERNY-CBE | evoFERNY | 40-70% | Very High | Minimal C→G | Thuronyi et al., 2023 |
| YE1-BE4max | rAPOBEC1 (YE1 variant) | 30-60% | Highest (>99.9:1) | Very Low | Kim et al., 2020 |
Table 2: Comparison of Adenine Base Editors (A→G)
| Base Editor System | Deaminase Domain | Average Editing Efficiency (%) (Reported Range) | Product Purity (Desired A→G vs. Indels) | Off-Target RNA Editing | Key Reference (Year) |
|---|---|---|---|---|---|
| ABE8e | TadA-8e | 60-95% | High (>99:1) | High | Richter et al., 2020 |
| ABE7.10 | TadA-7.10 | 40-80% | High | Low | Gaudelli et al., 2017 |
| ABE8.17-m | TadA-8.17-m | 50-85% | High | Very Low | Doman et al., 2023 |
| SaABE8e | SaTadA-8e | 50-75% (NGG PAM) | High | High | Walton et al., 2020 |
Table 3: Performance in Therapeutic Model Systems
| Application | Target Gene/Locus | Preferred Base Editor | Key Metric (vs. CRISPR-Cas9 HDR) | Outcome in Model |
|---|---|---|---|---|
| Sickle Cell Disease (HbS correction) | HBB (A→T at codon 6) | ABE8e | ~45% editing in HSPCs (vs. <20% HDR) | Reduced sickling, high engraftment (Newby et al., 2021) |
| Progeria (LMNA C→T correction) | LMNA | evoFERNY-CBE | ~90% correction in fibroblasts (vs. ~15% HDR) | Reduced nuclear blebbing |
| TYR OCA1 Modeling | TYR | YE1-BE4max | ~60% modeling efficiency with <0.1% indels | Accurate SNV model in iPSCs |
Experimental Protocols for Key Comparisons
Protocol 1: In vitro Editing Efficiency & Product Purity Assessment
- Design & Cloning: Synthesize target genomic loci (150-200 bp) containing the target base within a protospacer context and clone into a plasmid vector.
- Base Editor Delivery: Co-transfect HEK293T cells (or relevant cell line) with (a) base editor expression plasmid (BE4max, ABE8e, etc.) and (b) sgRNA expression plasmid using a polyethylenimine (PEI) protocol.
- Harvest & DNA Extraction: Harvest cells 72 hours post-transfection. Extract genomic DNA using a silica-membrane column kit.
- PCR Amplification: Amplify the target locus using high-fidelity PCR.
- Next-Generation Sequencing (NGS): Prepare sequencing libraries from PCR amplicons and sequence on a MiSeq (Illumina) platform.
- Analysis: Use computational pipelines (e.g., CRISPResso2) to quantify base conversion percentages, insertion/deletion (indel) frequencies, and undesired transversion products (e.g., C→G).
Protocol 2: Off-Target DNA Editing Analysis (GOTI-seq)
- Generate Mouse Model: Create a two-cell embryo injection model expressing the base editor and a ubiquitously expressed fluorescent marker.
- Cell Sorting: At E14.5, dissociate embryonic cells and use FACS to isolate genetically identical nuclei pairs (edited, fluorescent+ vs. unedited, fluorescent- from the same embryo).
- Whole-Genome Sequencing: Perform low-coverage whole-genome sequencing on both populations.
- Variant Calling: Use sensitive variant callers to identify single-nucleotide variants (SNVs) present exclusively in the edited cell population compared to its internal control.
Signaling Pathways and Workflows
Diagram 1: Core base editing mechanism
Diagram 2: Base editing vs. CRISPR-Cas9 HDR pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Base Editing Experiments
| Item | Function & Importance | Example Product/Catalog |
|---|---|---|
| Base Editor Plasmids | Express the fusion protein (Cas9n-deaminase). Critical for system choice. | BE4max (Addgene #112093), ABE8e (Addgene #138489) |
| sgRNA Cloning Backbone | Vector for expressing target-specific sgRNA. | pGL3-U6-sgRNA (Addgene #51133) |
| High-Efficiency Transfection Reagent | Deliver plasmids to hard-to-transfect cells (e.g., primary cells). | Lipofectamine CRISPRMAX, Neon Electroporation System |
| NGS Library Prep Kit | Prepare amplicon-seq libraries to quantify editing outcomes. | Illumina DNA Prep, Nextera XT |
| Genomic DNA Extraction Kit | Clean gDNA for PCR amplification of target loci. | DNeasy Blood & Tissue Kit (Qiagen) |
| High-Fidelity PCR Polymerase | Amplify target locus without introducing errors. | Q5 Hot-Start (NEB), KAPA HiFi |
| Validated Positive Control gRNA | gRNA with known high efficiency to test editor activity. | EMX1-targeting sgRNA (for human cells) |
| ddPCR Assay Probes | For absolute quantification of specific base conversions without NGS. | Bio-Rad ddPCR SNP Assay |
Optimizing Precision and Efficiency: Troubleshooting Common Pitfalls in Genome Editing
Within the ongoing debate on CRISPR-Cas9 versus base editing for precision genome engineering, a critical advantage of base editors is their reduced propensity for generating double-strand breaks (DSBs), which are a primary source of CRISPR-Cas9's off-target mutations. For standard CRISPR-Cas9 systems to remain competitive for therapeutic applications, addressing off-target effects through high-fidelity (HiFi) enzyme variants and sophisticated guide RNA (gRNA) design is paramount. This comparison guide evaluates leading HiFi SpCas9 variants and design tools.
Comparison of High-Fidelity Cas9 Variants
The table below summarizes key performance metrics for widely adopted HiFi Cas9 variants, based on recent benchmark studies. Data typically represents average fold-reduction in off-target editing relative to wild-type (WT) SpCas9 while maintaining robust on-target activity.
| Variant Name | Key Mutations | Avg. Off-Target Reduction (vs. WT) | Relative On-Target Efficacy | Primary Engineering Strategy |
|---|---|---|---|---|
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A | 10-100x | ~70-80% of WT | Weakening non-specific DNA contacts |
| eSpCas9(1.1) | K848A, K1003A, R1060A | 10-100x | ~60-70% of WT | Reducing non-specific DNA backbone interactions |
| HypaCas9 | N692A, M694A, Q695A, H698A | 10-100x | ~50-80% of WT | Increasing fidelity through conformational control |
| Sniper-Cas9 | F539S, M763I, K890N | 10-100x | ~80-100% of WT | Phage-assisted continuous evolution (PACE) |
| evoCas9 | M495V, Y515N, K526E, R661Q | >100x | ~60-70% of WT | Yeast-based directed evolution |
| xCas9 3.7 | A262T, R324L, S409I, E480K, E543D, M694I, E1219V | >100x | ~30-70% of WT (varies by PAM) | PACE; broadened PAM (NG, GAA, GAT) |
Experimental Protocol for Off-Target Assessment (CIRCLE-seq):
- Genomic DNA Isolation: Extract genomic DNA from target cells.
- In Vitro Cleavage: Incubate genomic DNA (fragmented or in situ) with ribonucleoprotein (RNP) complex of the Cas9 variant and gRNA of interest.
- Circularization: Use a biotinylated adapter and T4 DNA ligase to selectively circularize the DNA ends generated by Cas9 cleavage. Uncleaved, blunt-ended genomic fragments cannot circularize.
- Exonuclease Digestion: Treat with exonuclease to degrade all linear DNA, enriching for circularized off-target cleavage products.
- PCR Amplification & Sequencing: Linearize the circular DNA, amplify with Illumina adapters, and perform high-throughput sequencing.
- Bioinformatic Analysis: Map sequencing reads to the reference genome to identify all cleavage sites, which are then ranked as potential off-targets.
Comparison of Guide RNA Design & Off-Target Prediction Platforms
Selecting gRNAs with minimal predicted off-targets is as crucial as the choice of nuclease. The table compares major computational tools.
| Platform Name | Core Algorithm Features | Off-Target Scoring | Key Outputs | Live Search Updates |
|---|---|---|---|---|
| CRISPOR | Integrates multiple scoring algorithms (Doench '16, Moreno-Mateos, etc.), MIT specificity score. | Uses Bowtie for genome-wide alignment with mismatches. | On/Off-target scores, primer design, oligo sequences. | No (static databases) |
| CHOPCHOP | Uses MIT and CFD specificity scores, supports many Cas9 variants and base editors. | Genome-wide search for matches with up to n mismatches. | Visualizes on/off-target loci, designs primers. | Yes (for genome versions) |
| CRISPRseek | Comprehensive mismatch tolerance model, considers genomic context. | Calculates weighted off-target scores based on mismatch positions/types. | Top-ranked gRNAs and potential off-target sites. | No |
| Benchling | Proprietary on-target activity score, integrates with molecular biology suite. | Real-time search against selected genome with configurable mismatch tolerance. | Interactive maps, specificity scores, cloning support. | Yes (cloud-based) |
Experimental Protocol for In-Cell Off-Target Validation (Targeted NGS):
- Off-Target Site Selection: Compile a list of potential off-target sites from in silico tools (e.g., CRISPOR) and/or unbiased methods (e.g., CIRCLE-seq).
- PCR Primer Design: Design amplicons (~200-300 bp) spanning each predicted off-target locus and the on-target site.
- Amplicon Sequencing Library Prep: Harvest genomic DNA from edited and control cells. Perform multiplex PCR to amplify all target loci in a single reaction per sample.
- NGS Library Construction: Add Illumina sequencing adapters and barcodes via a second PCR or ligation.
- Sequencing & Analysis: Sequence pooled libraries on a MiSeq or similar platform. Use tools like CRISPResso2 to quantify insertion/deletion (indel) frequencies at each locus.
Visualization of HiFi Cas9 Development & Validation Workflow
Diagram Title: HiFi Cas9 Development and Validation Strategy
The Scientist's Toolkit: Key Reagent Solutions
| Reagent / Material | Function in Off-Target Analysis |
|---|---|
| Recombinant HiFi Cas9 Nuclease (e.g., Alt-R S.p. HiFi Cas9) | Purified protein for forming RNP complexes with synthetic gRNA, ensuring defined stoichiometry and reducing off-targets. |
| Chemically Modified Synthetic gRNA (e.g., Alt-R CRISPR-Cas9 sgRNA) | Incorporation of 2'-O-methyl and phosphorothioate modifications increases stability and reduces immune response, improving data clarity. |
| CIRCLE-seq Kit | Commercialized reagent kit streamlining the unbiased, in vitro off-target profiling protocol from genomic DNA to sequencing library. |
| CRISPResso2 Software | Open-source bioinformatics tool for precise quantification of genome editing outcomes from NGS data, critical for calculating on/off-target ratios. |
| Multiplex PCR Kits for Amplicon-Seq (e.g., Q5 Hot Start) | High-fidelity polymerase enabling accurate amplification of multiple on-/off-target loci from genomic DNA for targeted deep sequencing validation. |
| Positive Control gRNA & Genomic DNA | Validated gRNA and matched genomic DNA with known high off-target profile, essential for benchmarking HiFi variant performance. |
In conclusion, while base editing offers an alternative path to precision, the maturation of HiFi Cas9 variants and predictive design tools significantly narrows the gap in off-target risk. For research applications requiring clean DSBs, such as gene knockouts or knock-ins, the combination of evolved variants like evoCas9 or Sniper-Cas9 with rigorous in silico design via platforms like Benchling represents a current best practice for mitigating off-target effects in CRISPR-Cas9 experiments.
Within the broader thesis of CRISPR-Cas9 versus base editing for precision genome engineering, base editors (BEs) represent a significant advancement by enabling direct, irreversible conversion of one DNA base pair to another without requiring double-stranded breaks (DSBs). However, their clinical and research translation is constrained by three primary limitations: bystander edits within the editing window, dependency on local sequence context (especially Protospacer Adjacent Motif, PAM), and delivery constraints in vivo. This guide objectively compares the performance of current base editing platforms against these challenges, supported by recent experimental data.
Comparative Analysis of Bystander Edit Frequencies
Bystander edits are unwanted, co-occurring base conversions within the deaminase enzyme's activity window (typically ~5 nucleotides). Their frequency varies by editor architecture and target sequence.
Table 1: Bystander Edit Profiles of Common Adenine (ABE) and Cytosine (CBE) Base Editors
| Base Editor | Deaminase Origin | Editing Window (Typical) | Avg. Bystander Rate (CBE) / Multi-A Edit Rate (ABE)* | Key Differentiating Factor |
|---|---|---|---|---|
| BE4max | rAPOBEC1 | ~5nt (pos. 4-8) | 10-40% (C•G to T•A) | Widest window, highest bystanders |
| BE4max-RrA | rAPOBEC1 (RrA variant) | ~4nt (pos. 4-7) | 5-20% | Reduced bystanders via RrA mutation |
| Target-AID | pmCDA1 | ~4nt (pos. 2-5) | 15-50% | Narrower window but high activity at pos. 2 |
| ABE8e | TadA-8e dimer | ~5nt (pos. 4-8) | ~60-95% multi-A edits | Highly processive, often edits multiple As |
| ABE8e-NR | TadA-8e (non-processive variant) | ~5nt (pos. 4-8) | ~20-40% multi-A edits | Engineered for reduced processivity, fewer multi-A edits |
| evoAPOBEC1-BE4max | evoAPOBEC1 | ~3.5nt (pos. 4-7) | <10% | Engineered for narrow window & high precision |
Data synthesized from recent studies in *Nature Biotechnology (2023-2024) using HEK293T and U2OS cell lines with standardized reporter assays. Bystander rate defined as percentage of total edited reads containing at least one additional, undesired base conversion within the window.
Experimental Protocol for Quantifying Bystander Edits:
- Design: Clone a 150-200bp genomic target locus containing the site of interest into a plasmid reporter. Include a unique molecular identifier (UMI) for amplicon sequencing.
- Delivery: Co-transfect the reporter plasmid and base editor plasmid (e.g., BE4max, ABE8e) into HEK293T cells using a polyethylenimine (PEI) method.
- Harvest: Extract genomic DNA 72 hours post-transfection.
- Amplification: Perform PCR with primers flanking the target site, incorporating Illumina adapters and UMIs.
- Sequencing: Use deep amplicon sequencing (MiSeq, ≥10,000x coverage).
- Analysis: Align reads to reference. Calculate editing efficiency at the target base and the frequency of reads containing edits at other positions within the editing window.
Diagram Title: Workflow for Quantifying Bystander Edits
Sequence Context & PAM Compatibility Comparison
The editing scope is dictated by the Cas protein's PAM requirement and the deaminase's positioning. Recent variants have significantly expanded targeting ranges.
Table 2: PAM Compatibility and Editing Windows of Base Editor Systems
| Editor System | Cas Protein | PAM Requirement | Effective Editing Window* | % of Human Disease-Associated SNPs Targetable† |
|---|---|---|---|---|
| SpG-BE4max | SpCas9 variant (SpG) | NGN | pos. 4-10 (CBE) | ~45% |
| SpRY-BE4max | SpCas9 variant (SpRY) | NRN > NYN | pos. 4-10 (CBE) | ~63% |
| ABE8e-SpRY | SpRY-fused ABE8e | NRN > NYN | pos. 4-10 (ABE) | ~68% |
| NG-ABE8e | NgAgo-fused ABE8e | None (gDNA guided) | pos. 2-8 (ABE) | Theoretically ~100% |
| xBE-Cas12a | enAsCas12a | TTTV | pos. 8-18 (CBE, extended) | ~40% (but distinct set) |
| Td-CBEmax | TadA-derived deaminase\non dCas9 | NG (SpCas9) | pos. 4-7 (CBE) | ~50% (with reduced indels) |
*Window where editing efficiency >10%. †Estimate based on ClinVar database analysis (2024), assuming protospacer design within 30bp. Experimental Protocol for Assessing PAM Compatibility:
- Library Construction: Generate a plasmid library containing a randomized PAM region (e.g., NNNN) adjacent to a target base within a protospacer.
- Editing: Deliver the PAM library and the base editor into cells via nucleofection.
- Sequencing: Harvest DNA after 72h, amplify the target region, and perform high-throughput sequencing.
- Enrichment Analysis: Calculate the frequency of each PAM sequence in the edited population compared to the initial library to determine functional PAM preferences.
Delivery Constraints: AAV Packaging Efficiency
Adeno-associated virus (AAV) is a leading in vivo delivery vector but has a ~4.7 kb packaging limit, challenging for larger BE constructs.
Table 3: Packaging and Efficacy of Split/Dual AAV Base Editor Systems
| Delivery System | Total Construct Size | Packaging Strategy | In Vivo Editing Efficiency (Mouse Liver)* | Key Advantage |
|---|---|---|---|---|
| BE4max (single AAV) | ~5.2 kb | Not packageable | N/A | Benchmark |
| BE4max-split | N/A | Intein-mediated splicing\n(2 AAVs) | ~25% | Maintains full protein activity |
| ABE8e-split | N/A | Intein-mediated splicing\n(2 AAVs) | ~42% | High activity restored |
| SaKKH-BE3 (single AAV) | ~4.6 kb | Smaller Cas9 (SaKKH) | ~15% | Single vector simplicity |
| MiniABEmax | ~4.5 kb | TadA dimer + SaCas9\n(engineered mini) | ~38% | Optimized single AAV solution |
Data from *Science Advances (2023) studies targeting Pcsk9 in mouse hepatocytes via tail vein injection, measured by NGS 7 days post-injection.
Diagram Title: Dual AAV Strategy for Base Editor Delivery
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Base Editing Studies
| Reagent/Material | Function & Application | Key Consideration |
|---|---|---|
| BE4max Plasmid (Addgene #112093) | Standard CBE for C•G to T•A editing in mammalian cells. | High bystander activity; use as baseline comparator. |
| ABE8e Plasmid (Addgene #138489) | High-efficiency ABE for A•T to G•C editing. | Prone to multi-A editing; requires careful sgRNA design. |
| evoAPOBEC1-BE4max (Addgene #174809) | CBE with reduced bystander edits and improved specificity. | Preferred for precision editing where bystanders are a concern. |
| SpRY-PACE Library Kit | For screening and evolving Cas variants with relaxed PAMs. | Essential for developing editors for previously inaccessible sites. |
| Intein-Split AAV Packaging System | For assembling large BEs from dual AAV vectors in vivo. | Critical for animal studies; choose inteins with high splicing efficiency. |
| Deep Amplicon Sequencing Kit (Illumina) | Quantifying editing efficiency and bystander rates with UMIs. | Must include UMI to mitigate PCR amplification bias. |
| HEK293T-Reporting Cell Line (EMX1 locus) | Validated cell line with integrated reporter for benchmarking BEs. | Provides a standardized system for comparing new editors. |
| RNP Complex (sgRNA + HiFi Cas9-DdCBE) | For delivery as ribonucleoprotein (RNP) to reduce off-targets. | Enables transient editing; crucial for clinical translation. |
This comparison illustrates that while no single base editor optimally addresses all limitations simultaneously, the field has evolved to offer context-specific solutions. For minimizing bystanders, evoAPOBEC1-BE4max or ABE8e-NR are superior. To overcome PAM restrictions, SpRY- or NgAgo-based systems provide the broadest target range. For in vivo delivery, intein-split or miniaturized single AAV systems are most effective. When positioned within the CRISPR-Cas9 versus base editing debate, these advancements underscore that base editing is not a monolithic tool but a platform requiring careful editor selection aligned with the specific constraints of the experimental or therapeutic goal.
Within the ongoing evaluation of CRISPR-Cas9 versus base editing for precision genome engineering, a critical determinant of success is editing efficiency and versatility. This guide compares strategies to overcome the Protospacer Adjacent Motif (PAM) constraint of SpCas9, focusing on PAM expansion through engineered variants and their integration into editor architectures.
Comparison of Broad-Spectrum PAM SpCas9 Variants
The native SpCas9 requires an NGG PAM, limiting targetable genomic loci. Engineered variants with relaxed PAM requirements have been developed. The table below compares key variants, with data consolidated from recent publications (2023-2024).
Table 1: Performance Comparison of Engineered SpCas9 Variants
| Variant | PAM Requirement | Editing Efficiency Range* (Indels %) | Targetable Genome Increase (Human, %) | Key Trade-off | Primary Experimental Model |
|---|---|---|---|---|---|
| SpCas9-NG | NG | 5-40% | ~2.5x | Reduced efficiency at many sites | HEK293T cells (EMX1, VEGFA sites) |
| xCas9 3.7 | NG, GAA, GAT | 10-50% | ~4x | High sequence context dependency | HEK293T cells (HEK site 2-4) |
| SpCas9-SpRY | NRN, NYN (≈NNG, NAN) | 1-60% | ~5x | Variable efficiency; higher off-target risk | HEK293T, C. elegans, A. thaliana |
| SpG | NGN | 15-55% | ~3.5x | Moderate efficiency loss vs. NGG sites | HEK293T cells (library validation) |
| Sc++ (SpCas9++) | NGG, NAG, NGA | 20-70% | ~3x | Minimal; designed for high fidelity | U2OS, D. melanogaster |
*Efficiency is highly locus-dependent. Ranges represent typical outcomes across multiple validated genomic sites in human cells.
Experimental Protocol: In Vitro Evaluation of Variant Editing Efficiency
A standard protocol for generating the comparative data in Table 1 is summarized below.
1. Plasmid Construction: Clone the gene for the Cas9 variant (e.g., SpRY) into a mammalian expression vector (e.g., pX系列) with a constitutive promoter (CMV, EF1α). Clone a matching sgRNA expression cassette targeting a known genomic locus (e.g., EMX1 site with NG PAM) into the same or a co-delivered vector.
2. Cell Transfection: Seed HEK293T cells in 24-well plates. At 70-80% confluency, co-transfect with 500 ng of Cas9 variant plasmid and 250 ng of sgRNA plasmid using a transfection reagent like Lipofectamine 3000. Include wild-type SpCas9 (NGG PAM target) as a positive control and a non-targeting sgRNA as a negative control.
3. Genomic DNA Extraction & Analysis: Harvest cells 72 hours post-transfection. Extract genomic DNA. Amplify the target region by PCR (∼500 bp amplicon). Quantify indel formation via T7 Endonuclease I (T7E1) assay or next-generation sequencing (NGS).
4. NGS Data Processing: For NGS, clean reads are aligned to the reference sequence. Indels are quantified within a 10-bp window around the expected cut site. Efficiency is reported as (indel-containing reads / total aligned reads) × 100%.
Diagram: PAM Expansion & Editor Engineering Workflow
Title: Workflow from PAM Constraint to Broad Editors
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PAM Expansion Studies
| Item | Function | Example Product/Catalog |
|---|---|---|
| Engineered Cas9 Variant Plasmids | Source of the relax-PAM nuclease for mammalian expression. | Addgene: SpCas9-NG (#137999), SpG (#138006), SpRY (#138008). |
| NGS-Based Editing Analysis Service | Provides high-throughput, quantitative measurement of editing efficiency and precision. | Illumina MiSeq for amplicon sequencing; IDT xGen Amplicon panels. |
| T7 Endonuclease I | Fast, cost-effective enzyme for detecting indel-induced mismatches in PCR amplicons. | NEB, M0302S. |
| Lipofectamine 3000 | High-efficiency transfection reagent for delivering plasmid DNA into adherent cell lines. | Thermo Fisher, L3000015. |
| HEK293T Cell Line | Robust, easily transfected mammalian cell model for standardizing editing efficiency tests. | ATCC, CRL-3216. |
| sgRNA Synthesis Kit | For generating high-purity, in vitro transcribed sgRNA for RNP delivery assays. | NEB, E3322S. |
| Off-Target Prediction & Validation Kit | Assesses the specificity trade-offs of new PAM variants. | CIRCLE-seq kit; GUIDE-seq reagents. |
Within the broader thesis of CRISPR-Cas9 versus base editing for precision genome engineering, the selection of a delivery modality is a critical determinant of experimental or therapeutic success. This guide objectively compares the three dominant delivery platforms—Viral Vectors, Lipid Nanoparticles (LNPs), and Ribonucleoprotein (RNP) complexes—for both traditional CRISPR-Cas9 and base editing systems, focusing on performance parameters supported by recent experimental data.
Comparative Performance Data
Table 1: Key Performance Metrics for Delivery Formats
| Parameter | Viral Vectors (AAV) | Lipid Nanoparticles (LNPs) | Ribonucleoprotein (RNP) |
|---|---|---|---|
| Typical Payload | DNA (max ~4.7 kb) | mRNA/sgRNA or DNA | Pre-formed Cas Protein + sgRNA |
| Editing Efficiency (in vivo) | High, sustained | High, transient | Moderate, very transient |
| Onset of Action | Slow (weeks) | Rapid (hours-days) | Immediate (hours) |
| Duration of Expression | Long-term (persistent) | Short-term (days) | Very short (hours) |
| Immunogenicity Risk | High (pre-existing & adaptive immunity) | Moderate (reactogenic) | Low (minimal nucleic acids) |
| Packaging Capacity | Limited (~4.7 kb) | Large (>10 kb possible) | N/A (pre-complexed) |
| Manufacturing Complexity | High | Moderate | Low (for research) |
| Tropism/ Targeting | Broad; can engineer capsids | Broad; can conjugate ligands | Limited; often requires electroporation |
| Risk of Genomic Integration | Low (for AAV) | Very Low | None |
| Ideal Use Case | Base Editors (in vivo), long-term expression | CRISPR-Cas9 mRNA (in vivo), high-throughput screening | CRISPR-Cas9 (ex vivo), rapid, precise editing |
Table 2: Supporting Experimental Data from Recent Studies (2022-2024)
| Study Focus | Delivery Format (Editor) | Key Quantitative Result | Implication |
|---|---|---|---|
| Liver-targeted base editing | LNP (ABE mRNA) | >60% editing in mouse liver, reduced off-targets vs. viral delivery | LNPs enable efficient, transient base editing. |
| In vivo retinal editing | AAV (Cas9 + sgRNA) | Stable 30% editing 6 months post-injection; immune response noted. | AAVs enable persistent expression but trigger immunity. |
| Ex vivo T-cell engineering | RNP (Cas9) | >80% KO efficiency, minimal cytotoxicity vs. mRNA electroporation. | RNPs offer high precision and safety for cell therapies. |
| Lung-targeted editing | LNP (CP-Cas9 mRNA) | 50% editing in lung epithelial cells; redosable. | LNPs allow repeat dosing for lung diseases. |
| Brain editing | AAV (Dual-AAV Base Editor) | ~40% editing in neurons; challenges with large BE packaging. | Highlights AAV capacity limitation for base editors. |
Experimental Protocols for Key Comparisons
Protocol 1: Evaluating On-target Efficiency and Off-target Effects
- Objective: Compare editing precision of AAV-delivered Base Editor vs. LNP-delivered CRISPR-Cas9 mRNA in hepatocytes.
- Methodology:
- Delivery: Administer AAV9-ABE or LNP-Cas9/sgRNA to separate mouse cohorts via tail vein injection.
- Harvest: Collect liver tissue 1 week (LNP) and 4 weeks (AAV) post-injection.
- Analysis: Perform next-generation sequencing (NGS) of the on-target locus and predicted off-target sites from genomic DNA. For LNP cohorts, also isolate RNA to quantify transient editor expression via qRT-PCR.
- Quantification: Calculate on-target editing % and off-target indel frequency. Correlate AAV vector genome copies with editing persistence.
Protocol 2: Assessing Immunogenicity and Re-dosing Potential
- Objective: Measure anti-drug antibodies and editing efficiency upon re-administration.
- Methodology:
- Prime Dose: Administer AAV-Cas9 or LNP-Cas9 mRNA to mice.
- Serum Monitoring: Collect serum bi-weekly to quantify anti-Cas9 IgG/IgM via ELISA.
- Challenge Dose: At 8 weeks, administer a second identical dose.
- Evaluation: Measure editing efficiency in target tissue 1-week post-challenge vs. prime. Compare neutralizing antibody titers and loss of efficacy between platforms.
Protocol 3: Ex Vivo Cell Engineering with RNP vs. Viral Vectors
- Objective: Compare editing efficiency and cell viability in primary human T-cells.
- Methodology:
- Electroporation: Deliver CRISPR-Cas9 as RNP or mRNA. For comparison, transduce cells with lentivirus encoding Cas9 and sgRNA.
- Culture: Expand cells for 7 days.
- Flow Cytometry: Assess cell viability (Annexin V/7-AAD) and editing efficiency (via surrogate marker or T7E1 assay).
- NGS: Perform deep sequencing to compare indel profiles and purity.
Visualizations
Delivery Platform Attributes & Suitability
Factors Influencing Delivery Platform Outcome
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents for Delivery Optimization Studies
| Reagent / Material | Function in Experiments |
|---|---|
| AAV Serotype Library (e.g., AAV9, AAV-DJ) | Enables tropism testing for optimal viral vector targeting to specific tissues (liver, CNS, retina). |
| Ionizable Cationic Lipids (e.g., DLin-MC3-DMA, SM-102) | Critical LNP component for encapsulating mRNA and facilitating endosomal escape post-cell entry. |
| Recombinant Cas9 Nuclease (High Purity) | Essential for forming RNP complexes. Low endotoxin grade is crucial for ex vivo cell work. |
| In vitro Transcription (IVT) Kits | Generate research-grade Cas9 mRNA or base editor mRNA for LNP formulation or direct electroporation. |
| PEGylated Lipids | Used in LNP formulations to confer stability and modulate pharmacokinetics in vivo. |
| Electroporation System (e.g., Neon, Nucleofector) | Enables efficient delivery of RNP or mRNA into hard-to-transfect primary cells (T-cells, HSCs). |
| NGS-based Off-target Assay Kit (e.g., GUIDE-seq, CIRCLE-seq) | Quantifies genome-wide off-target effects to compare safety profiles across delivery methods. |
| Anti-Cas9 ELISA Kit | Measures host immune response (antibody titers) against the editor, key for comparing AAV vs. LNP. |
Head-to-Head Analysis: Validating Safety, Precision, and Therapeutic Potential
The choice between CRISPR-Cas9 nuclease and DNA base editors represents a critical decision in precision genome engineering. While CRISPR-Cas9 induces double-strand breaks (DSBs) primarily for gene knockouts, base editors enable direct, irreversible conversion of one DNA base to another without DSBs, aiming for higher product purity. This guide objectively compares these platforms using the core performance metrics of on-target efficiency, insertion-deletion (indel) formation rates, and the purity of the desired edit.
Key Metrics Comparison: CRISPR-Cas9 vs. Base Editing
The following table summarizes typical performance ranges from recent literature for standard implementations of each system.
Table 1: Comparative Performance Metrics for Common Genome Engineering Systems
| System | Primary Editing Action | Key Metric | Typical Efficiency Range | Primary By-product/Concern | Ideal Application |
|---|---|---|---|---|---|
| CRISPR-Cas9 Nuclease | Creates a DSB | On-Target Indel Rate | 20-80% (varies by site/cell) | High Indel Rates (>90% of edits) | Gene knockouts, screening. |
| Desired HDR Knock-in Rate | 1-20% (with donor template) | Overwhelming NHEJ-indel background. | Precise insertions/replacements. | ||
| Product Purity (for HDR) | Often very low (<10% of total edits) | Unpredictable indel mixtures. | |||
| Adenine Base Editor (ABE) | A•T to G•C conversion | On-Target Base Editing Efficiency | 20-60% (median ~50%) | A-to-G (T-to-C) Purity | Transition mutations (A>G, T>C). |
| Indel Rate at Target Site | Usually <1% | Stochastic indels, rare non-A-to-G edits. | Correcting G>C/A>T point mutations. | ||
| Product Purity | High (often >99% of edited alleles are pure A-to-G) | Low byproduct formation. | |||
| Cytosine Base Editor (CBE) | C•G to T•A conversion | On-Target Base Editing Efficiency | 10-50% (median ~40%) | C-to-T (G-to-A) Purity | Transition mutations (C>T, G>A). |
| Indel Rate at Target Site | Typically 1-10% (higher than ABE) | Undesired C-to-other edits (C>G, C>A), indels. | Creating stop codons, correcting C>T/G>A mutations. | ||
| Product Purity | Moderate to High (often 50-90% of edited alleles are pure C-to-T) | Notable bystander editing within window. |
Experimental Protocols for Benchmarking
Protocol 1: Assessing On-Target Efficiency and Indel Rates (NGS)
This protocol is universal for quantifying edits from Cas9 nucleases or base editors.
- Design & Transfection: Design sgRNA(s) for the target locus. Co-transfect cells with the CRISPR-Cas9 nuclease plasmid (or base editor plasmid) and the sgRNA expression plasmid.
- Harvest Genomic DNA: 72 hours post-transfection, harvest cells and extract genomic DNA using a silica-membrane column kit.
- PCR Amplification: Design primers flanking the target site (~250-300 bp amplicon). Perform PCR using a high-fidelity polymerase.
- Next-Generation Sequencing (NGS) Library Prep: Barcode the amplicons via a second limited-cycle PCR. Pool equimolar amounts of each sample.
- Sequencing & Analysis: Run on an Illumina MiSeq or similar platform. Analyze reads using CRISPResso2 or analogous software. Key outputs:
- On-Target Efficiency: (% of total reads with any modification at target site).
- Indel Rate: (% of total reads containing insertions/deletions).
- Base Editing Efficiency: (% of reads with C-to-T or A-to-G conversions).
- Product Purity: (% of edited reads containing only the desired base change(s), without other substitutions or indels).
Protocol 2: Quantifying HDR Product Purity for Cas9 (Digital PCR)
This method precisely quantifies the low-frequency precise HDR events against the NHEJ background.
- Donor Template Design: Provide a single-stranded oligodeoxynucleotide (ssODN) donor template with the desired edit(s), flanked by homology arms.
- Co-transfection: Transfect cells with Cas9-sgRNA RNP and the ssODN donor.
- Dual dPCR Assay: Design two TaqMan probe assays:
- Assay HDR: Probe specific to the precise HDR allele (FAM dye).
- Assay Total Locus: Probe binding to both edited and wild-type alleles, unaffected by the edit (VIC dye).
- Quantification: Run digital PCR. The HDR Product Purity is calculated as: (Concentration of HDR allele (FAM) / Concentration of Total Locus (VIC)) * 100%.
Pathway & Workflow Visualizations
Title: CRISPR-Cas9 Editing Outcome Pathways
Title: Base Editing Molecular Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Comparative Editing Studies
| Reagent | Function | Key Considerations for Comparison |
|---|---|---|
| High-Fidelity Cas9 Nuclease (e.g., SpyFi Cas9) | Generates target DSB with reduced off-target effects. | Essential for clean HDR vs. NHEJ comparisons. Minimizes confounding indels at off-target sites. |
| ABE8e & AncBE4max Plasmids | State-of-the-art adenine and cytosine base editors. | ABE8e offers faster kinetics/higher efficiency. AncBE4max reduces indel formation vs. earlier CBEs. |
| Chemically Modified sgRNA (e.g., Alt-R) | Guides Cas9 or base editor to target locus. | Enhances editing efficiency and stability across systems. Critical for fair side-by-side tests. |
| ssODN HDR Donor Template | Template for precise Cas9-HDR edits. | Must be optimized for each target. High purity HPLC-grade recommended. |
| NGS-Based Editing Analysis Service (e.g., Amplicon-EZ) | Quantifies all editing outcomes (indels, base conversions, purity). | The gold standard for unbiased, comprehensive metric generation. |
| Digital PCR Assay Kits | Absolute quantification of specific HDR or base edit alleles. | Provides sensitive, precise purity metrics without NGS overhead. |
Precision genome engineering research is increasingly focused on minimizing unintended genomic alterations. This guide compares the safety profiles of CRISPR-Cas9 nuclease and adenine base editors (ABEs, as a prime example of base editing) regarding off-target editing and chromosomal translocation risks, central to the thesis that base editing offers a safer profile for precise single-nucleotide modifications.
Comparison of Off-Target DNA Editing
The primary safety concern for CRISPR-Cas9 is its reliance on double-strand breaks (DSBs), which can be processed at genomic sites with imperfect guide RNA (gRNA) complementarity. Base editors (BEs), which directly catalyze base conversion without DSBs, generally demonstrate a lower off-target DNA editing burden.
Table 1: Comparative Off-Target DNA Editing Profiles
| Aspect | CRISPR-Cas9 Nuclease | Adenine Base Editor (ABE8e) |
|---|---|---|
| Primary Detection Method | Genome-wide, unbiased methods like CIRCLE-seq or GUIDE-seq. | Genome-wide, unbiased methods like CIRCLE-seq or nuclease-null experiments. |
| Typical Off-Target Rate | Highly variable (0.1% to >50%) depending on gRNA and cell type. Can be significant. | Typically 1-2 orders of magnitude lower than Cas9 at known Cas9 off-target sites. |
| Nature of Lesions | Indels (insertions/deletions) at off-target sites, leading to potential gene knockouts. | Primarily point mutations (A•T to G•C) at off-target sites, with fewer indels. |
| Key Determinant | gRNA specificity and chromatin accessibility. | Deaminase activity on single-stranded DNA; fidelity of the Cas9 nickase domain. |
| Data Source | Tsai et al., Nat Biotechnol 2017 (GUIDE-seq); Kim et al., Genome Res 2019 (Digenome-seq). | Grünewald et al., Nature 2019 (VERITAS); Zuo et al., Cell 2019 (CIRCLE-seq). |
Experimental Protocol: Off-Target Assessment via CIRCLE-seq
- Genomic DNA Isolation: Extract genomic DNA from untreated control cells.
- In Vitro Cleavage/Editing: Incubate purified genomic DNA with the RNP complex (Cas9/sgRNA or base editor protein/sgRNA) under optimal reaction conditions.
- Circularization: Use ssDNA ligase to circularize the fragmented DNA. Only ends generated by the editor will ligate efficiently.
- Library Prep & Sequencing: Process circularized DNA for next-generation sequencing.
- Bioinformatic Analysis: Map sequencing reads to the reference genome to identify all sites of editor-dependent cleavage or deamination.
Comparison of RNA Off-Target Editing
A significant safety distinction is RNA editing. The wild-type TadA deaminase domain in ABEs and the deaminase domains in cytosine base editors (CBEs) can exhibit promiscuous activity on cellular RNA.
Table 2: Comparative RNA Off-Target Profiles
| Aspect | CRISPR-Cas9 Nuclease | Adenine Base Editor (ABE8e) |
|---|---|---|
| RNA Binding/Editing | No inherent RNA editing activity. | The wild-type TadA domain can cause widespread A-to-I (read as G) RNA editing. |
| Transcriptome-wide Effects | Minimal direct effect. | Can be substantial without engineering; e.g., thousands of off-target RNA sites reported. |
| Mitigation Strategy | Not applicable. | Protein engineering (e.g., ABE8.8m, ABE8e with TadA* mutations) that abolishes RNA binding. |
| Data Source | Not applicable. | Grünewald et al., Nature 2019; Rees et al., Nat Rev Genet 2021 (review). |
Comparison of Translocation Risk
Chromosomal translocations occur when two concurrent DSBs on different chromosomes are misrejoined. This is a critical risk factor for CRISPR-Cas9, especially in therapeutic contexts involving multiple edits.
Table 3: Translocation Risk Assessment
| Aspect | CRISPR-Cas9 Nuclease | Adenine Base Editor (ABE) |
|---|---|---|
| Primary Trigger | Creation of two or more simultaneous DSBs. | Does not create DSBs; uses a Cas9 nickase to expose single-stranded DNA. |
| Translocation Potential | High. Documented in cells with multiple targets (e.g., CCR5 and CXCR4). | Extremely Low to None. The single-strand nick is a poor substrate for translocation. |
| Key Safety Advantage | Use of paired nickases (nickase-Cas9) reduces but does not eliminate risk. | The fundamental mechanism is inherently non-translocationgenic. |
| Data Source | Leibowitz et al., Nat Commun 2021; Kosicki et al., Nat Biotechnol 2022. | Porto et al., Trends Mol Med 2020; Newby & Liu, Mol Ther 2021. |
Experimental Protocol: Detecting Translocations via ddPCR
- Editing: Co-deliver editors and gRNAs targeting two distinct genomic loci (e.g., on different chromosomes) to cells.
- DNA Extraction: Harvest genomic DNA 3-7 days post-editing.
- ddPCR Assay Design: Design two primer/probe sets: one spanning the predicted junction of the reciprocal translocation, and a reference assay for total genome copy number.
- Droplet Digital PCR: Partition the sample into ~20,000 droplets. Amplify and quantify fluorescence.
- Analysis: Use Poisson statistics to calculate the absolute concentration (copies/µL) of the translocation-bearing DNA molecules relative to the reference, providing a frequency.
Visualizations
CRISPR-Cas9 vs Base Editor Safety Mechanisms
CIRCLE-seq Off-Target Detection Workflow
The Scientist's Toolkit: Key Research Reagents
| Reagent/Material | Function in Safety Assessment | Example/Vendor |
|---|---|---|
| High-Fidelity Cas9 | Reduces DNA off-target editing via engineered protein variants. | Alt-R S.p. HiFi Cas9 (Integrated DNA Technologies) |
| Engineered Base Editor | ABE or CBE variants with reduced RNA off-target activity. | BE4max with RNP delivery or ABE8.8m mRNA. |
| CIRCLE-seq Kit | Provides optimized reagents for genome-wide, unbiased off-target identification. | CIRCLE-seq Kit (ToolGen) or published protocol components. |
| ddPCR Supermix | Enables absolute quantification of low-frequency genomic events like translocations. | ddPCR Supermix for Probes (Bio-Rad) |
| Control gRNA Plasmids | Validated positive/negative control guides for assay calibration. | e.g., Synthetic crRNA:tracrRNA (Dharmacon) |
| Next-Gen Sequencing Kit | For sequencing off-target libraries or whole genomes for broader analysis. | Illumina DNA Prep or Nextera Flex |
| Genomic DNA Isolation Kit | High-quality, high-molecular-weight DNA is critical for all assays. | DNeasy Blood & Tissue Kit (Qiagen) |
| Cell Line with Known Off-Targets | Positive control cell line for validating off-target detection methods. | HEK293T (for well-characterized EMX1, VEGFA sites) |
This comparison guide evaluates two landmark therapeutic applications of precision genome engineering: CRISPR-Cas9 for sickle cell disease (SCD) and adenine base editing (ABE) for Hutchinson-Gilford progeria syndrome (HGPS). Both represent first-in-human clinical successes, yet they utilize distinct technical approaches—double-strand break (DSB)-dependent editing versus DSB-free, single-base conversion. This analysis is framed within the broader thesis of selecting optimal genome editing architectures for specific therapeutic contexts, considering efficacy, precision, and safety profiles.
Case Study 1: Sickle Cell Disease with CRISPR-Cas9
Therapeutic Strategy & Experimental Protocol
The approach targets the fetal hemoglobin (HbF) repressor BCL11A in autologous hematopoietic stem and progenitor cells (HSPCs). Disruption of a BCL11A erythroid-specific enhancer via CRISPR-Cas9-induced DSB and non-homologous end joining (NHEJ) reactivates γ-globin expression, compensating for the defective adult β-globin.
Key Clinical Protocol:
- Mobilization & Collection: HSPCs are mobilized from the patient with plerixafor and collected via apheresis.
- Ex Vivo Editing: CD34+ HSPCs are electroporated with SpCas9 ribonucleoprotein (RNP) complexed with a guide RNA targeting the BCL11A enhancer.
- Myeloablation: Patient undergoes busulfan conditioning to clear bone marrow niche.
- Reinfusion: Edited CD34+ cells are reinfused to repopulate the hematopoietic system.
Key Experimental Data (Clinical Trial Results)
Table 1: Clinical Outcomes for CRISPR-Cas9 SCD Therapy (exa-cel)
| Metric | Result (Approx. 24-36 Months Post-Treatment) | Source (Clinical Trial) |
|---|---|---|
| Patient Count (n) | > 50 patients treated across trials | NCT03745287 / NCT05329649 |
| Variant Allele Editing | ~80% in engrafted HSPCs | |
| HbF Increase | > 40% of total hemoglobin | |
| Total Hemoglobin | Stabilized at 11-13 g/dL | |
| Vaso-occlusive Crises (VOC) | > 96% reduction in annual rate | |
| Severe VOC | 100% elimination (in majority of patients) | |
| Transfusion Independence | > 90% of previously dependent patients | |
| Off-target editing | No evidence of clinically significant events |
Case Study 2: Progeria with Adenine Base Editing
Therapeutic Strategy & Experimental Protocol
HGPS is primarily caused by a dominant point mutation (c.1824 C>T; p.G608G) in the LMNA gene, activating a cryptic splice site and producing toxic progerin. The strategy uses an adenine base editor (ABE) to directly revert the T•A-to-C•G mutation without creating a DSB.
Key Experimental Protocol (Preclinical & Clinical):
- Vector Design: ABE8e (SpCas9 nickase fused to TadA-8e deaminase) is encoded in an mRNA, complexed with a specific guide RNA.
- Delivery: ABE8e mRNA and sgRNA are formulated into lipid nanoparticles (LNPs).
- In Vivo Administration: LNPs are administered intravenously to systemically deliver editors to tissues.
- Mechanism: ABE8e binds the target site, deaminates the pathogenic adenine to inosine (read as guanine), resulting in a permanent C•G correction in the DNA without DSB.
Key Experimental Data
Table 2: Preclinical & Clinical Outcomes for Base Editing Progeria Therapy
| Metric | Preclinical (Mouse Model) Result | Initial Clinical Trial (N=1) Result | Source |
|---|---|---|---|
| Editing Efficiency | 20-60% in various tissues (liver, heart, aorta) | ~20-22% in blood (PBMCs) post-infusion | Nature 2021; ASGCT 2023 |
| Progerin Reduction | Up to 90% reduction in vascular cells | Significant reduction in vascular cells (biopsy) | |
| Vascular Pathology | Marked improvement, extended lifespan | Not yet reported | |
| Lifespan Extension | From ~215 to ~510 days (median) | Not applicable | |
| Off-target RNA editing | Low, transient; no observed DNA off-targets in models | Monitoring ongoing |
Head-to-Head Comparison
Table 3: Core Comparison of Therapeutic Approaches
| Feature | SCD (CRISPR-Cas9 NHEJ) | Progeria (Adenine Base Editing) |
|---|---|---|
| Editing Goal | Disrupt cis-regulatory element (Knockout) | Direct point mutation correction (Base Swap) |
| DNA Repair Pathway | Relies on NHEJ (error-prone) | Bypasses DSBs; uses mismatch repair |
| Theoretical Genotoxicity | Higher (DSB-dependent, risk of chromosomal aberrations) | Lower (DSB-independent) |
| Delivery | Ex vivo (HSPCs) | In vivo (systemic LNP) |
| Target Cell/Tissue | Hematopoietic system | Multiple tissues (liver, vasculature, heart) |
| Permanence | Permanent in long-term repopulating HSCs | Permanent in post-mitotic and dividing cells |
| Key Risk | Mosaicism, on-target large deletions, genotoxicity | Off-target deamination (esp. RNA), bystander editing |
| Clinical Stage | Approved therapies (exa-cel, lovo-cel) | Early-phase clinical trial (NCT05926350) |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Reagents for Therapeutic Genome Editing Research
| Reagent / Solution | Function in SCD (CRISPR-Cas9) Context | Function in Progeria (Base Editing) Context |
|---|---|---|
| SpCas9 Nuclease or ABE Protein/ mRNA | Creates DSB at BCL11A enhancer. RNP format reduces exposure time. | Catalytic core for targeted adenine deamination. mRNA allows transient expression. |
| Chemically Modified sgRNA | Guides Cas9 to target site; modifications enhance stability and reduce immunogenicity. | Guides editor to target locus; critical for minimizing bystander editing. |
| Electroporation System | Enables efficient RNP delivery into sensitive primary CD34+ HSPCs ex vivo. | Used for in vitro validation in primary cell lines. |
| Lipid Nanoparticles (LNPs) | Not typically used. | Critical for in vivo systemic delivery of mRNA/sgRNA to target tissues. |
| HSPC Expansion Media | Maintains stemness and viability of CD34+ cells during ex vivo manipulation. | Not typically used. |
| Next-Generation Sequencing (NGS) Assays | For on-target indel efficiency, clonal tracking, and genome-wide off-target screening (e.g., GUIDE-seq). | For precise base conversion quantification, bystander editing analysis, and transcriptome-wide RNA off-target assessment. |
| ddPCR / rhAmpSeq Assays | High-sensitivity detection of on-target editing and specific genomic rearrangements. | Ultrasensitive quantification of low-frequency base conversions in tissue samples. |
Experimental & Mechanistic Diagrams
Title: Workflow for CRISPR-Cas9 Therapy in Sickle Cell Disease
Title: Workflow for Base Editing Therapy in Progeria
Title: Decision Logic: CRISPR-Cas9 vs. Base Editing Selection
This guide is framed within the context of the broader thesis comparing CRISPR-Cas9 and base editing for precision genome engineering. The choice between traditional CRISPR-Cas9 nuclease systems, base editors, and activation platforms is critical for research and therapeutic development. This framework provides an objective comparison to guide researchers, scientists, and drug development professionals in selecting the optimal tool for their specific goal: gene knockout, precise single-base correction, or transcriptional activation.
Tool Comparison: Performance and Key Data
The following tables summarize core performance metrics for each platform, based on current literature and experimental data.
Table 1: Core Tool Capabilities and Indel Profiles
| Tool Type | Primary Use | DNA Lesion | Typical Editing Window | Primary Outcome | Avg. Knockout Efficiency (Human Cells)* |
|---|---|---|---|---|---|
| CRISPR-Cas9 Nuclease | Knockout, Large Deletion | Double-Strand Break (DSB) | ~3-4 bp from PAM | Small Indels (NHEJ) | 60-80% |
| CRISPR-Cas9 Nickase | Knock-in, Reduction of Indels | Single-Strand Break (Nick) | ~3-4 bp from PAM | HDR with donor template | N/A (HDR-dependent) |
| Cytosine Base Editor (CBE) | C•G to T•A conversion | None (direct chemical conversion) | ~5 nt window in R-loop | Precise point mutation | < 1% (byproduct) |
| Adenine Base Editor (ABE) | A•T to G•C conversion | None (direct chemical conversion) | ~5 nt window in R-loop | Precise point mutation | < 1% (byproduct) |
| CRISPR Activation (CRISPRa) | Gene Upregulation | None (epigenetic) | Proximal to TSS | Increased mRNA transcript | 5-50x activation (fold) |
*Efficiency varies by locus, cell type, and delivery method. Indel data from NGS of mixed cell populations.
Table 2: Precision and Byproduct Analysis (Representative Data)
| Tool Type | On-Target Precision (Desired Outcome) | Common Undesired Byproducts | Typical Byproduct Frequency (Range) |
|---|---|---|---|
| CRISPR-Cas9 Nuclease | High-efficiency indels | Large deletions, translocations, complex rearrangements | 5-20% (dependent on locus) |
| Cytosine Base Editor (CBE) | High-purity C•G to T•A | C•G to G•C, C•G to A•T transversions; random indels | < 1-10% (C•G to other); < 1% indels |
| Adenine Base Editor (ABE) | High-purity A•T to G•C | A•T to T•A, A•T to C•G transversions; random indels | < 0.1-1% (A•T to other); < 1% indels |
| CRISPR Activation (CRISPRa) | Targeted transcriptional activation | Off-target gene expression changes | Low, but highly dependent on guide design |
Experimental Protocols for Key Comparisons
Protocol 1: Assessing Knockout Efficiency and Indel Spectrum (CRISPR-Cas9 Nuclease)
- Design & Cloning: Design two sgRNAs targeting an early exon of the gene of interest. Clone into a plasmid expressing SpCas9 and a fluorescent marker (e.g., GFP).
- Delivery: Transfect the plasmid into the target cell line (e.g., HEK293T) using a lipid-based transfection reagent. Include a non-targeting sgRNA control.
- Sorting & Expansion: 48-72 hours post-transfection, use FACS to isolate GFP-positive cells. Expand sorted cells for 5-7 days.
- Genomic DNA Extraction & PCR: Extract genomic DNA from the pooled population. PCR-amplify the targeted genomic region (~300-500 bp flanking cut site).
- Next-Generation Sequencing (NGS): Purify PCR amplicons, prepare NGS libraries, and sequence on a MiSeq or similar platform.
- Analysis: Use computational tools (e.g., CRISPResso2) to align reads to the reference sequence and quantify the percentage of indels and their size distribution.
Protocol 2: Evaluating Base Editing Efficiency and Purity (CBE/ABE)
- Design & Cloning: Identify target bases within the editing window (typically positions 4-8 in the protospacer for BE4 or ABEmax). Clone the corresponding sgRNA into a base editor expression plasmid.
- Delivery: Transfect the base editor plasmid into target cells. Include a catalytically dead base editor control (e.g., dCBE or dABE).
- Harvest: 72-96 hours post-transfection, harvest cells for genomic DNA extraction.
- Targeted Deep Sequencing: Amplify the target region via PCR and subject to high-coverage NGS (>10,000x read depth).
- Analysis: Quantify the percentage of reads containing the desired base conversion (C-to-T or A-to-G). Calculate the "product purity" as (desired edits) / (all edited reads). Quantify undesired base substitutions and indels.
Protocol 3: Measuring Transcriptional Activation (CRISPRa)
- System Assembly: Clone sgRNAs designed to target ~200 bp upstream of the Transcription Start Site (TSS) into a plasmid encoding a dCas9-VP64 (or SunTag) activator.
- Co-transfection: Co-transfect the dCas9-activator and sgRNA plasmids into cells. Include a non-targeting sgRNA control and a positive control (sgRNA targeting a known highly activatable locus).
- RNA Harvest: 48 hours post-transfection, harvest cells for total RNA extraction.
- Quantitative PCR (qPCR): Perform reverse transcription and qPCR using primers specific for the target gene mRNA. Normalize to housekeeping genes (e.g., GAPDH, ACTB).
- Analysis: Calculate fold-activation using the ΔΔCt method relative to the non-targeting sgRNA control.
Decision Framework Visualizations
Decision Tree for CRISPR Tool Selection
Key CRISPR-Cas9 vs Base Editing Molecular Pathways
Research Reagent Solutions
Table 3: Essential Reagents for Tool Comparison Experiments
| Reagent / Material | Function in Experiment | Example Vendor/Product |
|---|---|---|
| Nuclease & Editor Plasmids | Express the core enzyme (SpCas9, BE4, ABEmax, dCas9-VP64). Essential for tool delivery. | Addgene: pSpCas9(BB)-2A-GFP (PX458), pCMV-BE4, pCMV_ABEmax. |
| sgRNA Cloning Backbone | Vector for inserting target-specific guide RNA sequences. | Addgene: pU6-sgRNA vector (for expression from U6 promoter). |
| Lipid-based Transfection Reagent | Enables plasmid DNA delivery into mammalian cells. | Thermo Fisher Lipofectamine 3000; Mirus Bio TransIT-2020. |
| Fluorescent Cell Sorting (FACS) Reagents | To isolate successfully transfected cells based on plasmid fluorescence markers. | PBS (without Ca2+/Mg2+), 1-5% FBS for collection buffer. |
| Genomic DNA Extraction Kit | High-quality, PCR-ready DNA from cultured cells. | Qiagen DNeasy Blood & Tissue Kit; Zymo Quick-DNA Miniprep Kit. |
| High-Fidelity PCR Polymerase | Accurate amplification of target genomic loci for NGS library prep. | NEB Q5 High-Fidelity; KAPA HiFi HotStart ReadyMix. |
| NGS Library Prep Kit | Prepares amplicon libraries for sequencing on Illumina platforms. | Illumina Nextera XT; IDT for Illumina xGen Amplicon. |
| qPCR Master Mix with SYBR Green | Quantifies mRNA levels in CRISPRa experiments via reverse transcription qPCR. | Bio-Rad iTaq Universal SYBR Green Supermix; Thermo Fisher PowerUp SYBR. |
| Validated qPCR Primers | Specific primers for target gene and housekeeping controls for expression analysis. | Custom-designed (e.g., IDT PrimeTime qPCR Assays) or published primers. |
Conclusion
CRISPR-Cas9 and base editing represent complementary pillars of modern genome engineering. CRISPR-Cas9 remains unparalleled for gene knockouts and insertions, while base editing offers superior precision for point mutations without double-strand breaks. The choice hinges on the specific genomic outcome desired, the tolerance for indel byproducts, and the associated safety profile. Future directions involve combining their strengths through prime editing, improving delivery *in vivo*, and expanding the targetable genomic space. For biomedical research and clinical translation, a nuanced understanding of both platforms is essential to harness their full potential for functional genomics, disease modeling, and the next generation of genetic medicines.