Deconstructing Cas9: A Structural Guide to Its Domain Architecture and Functional Implications for Genome Engineering
This article provides a comprehensive structural and functional analysis of the Cas9 protein, the cornerstone enzyme of CRISPR-Cas9 genome editing.
Deconstructing Cas9: A Structural Guide to Its Domain Architecture and Functional Implications for Genome Engineering
Abstract
This article provides a comprehensive structural and functional analysis of the Cas9 protein, the cornerstone enzyme of CRISPR-Cas9 genome editing. Targeted at researchers and drug development professionals, it begins by deconstructing the fundamental domain architecture of Cas9, detailing the roles of the REC (recognition) and NUC (nuclease) lobes, HNH, and RuvC domains. The article then explores how this structural knowledge informs experimental methodologies, from sgRNA design to complex delivery systems. It further addresses common structural challenges and optimization strategies, including off-target effects and specificity enhancement. Finally, it validates these insights by comparing natural Cas9 orthologs (SpCas9, SaCas9) and engineered variants (high-fidelity, compact, PAM-relaxed), highlighting their distinct applications. The synthesis offers a roadmap for leveraging Cas9's structural blueprint to advance therapeutic development and precision genomic research.
The Structural Blueprint of Cas9: Decoding Domain Architecture and Catalytic Mechanics
The CRISPR-Cas9 system represents a paradigm shift in molecular biology, evolving from a prokaryotic adaptive immune mechanism to a programmable genome editing tool. This whitepaper examines Cas9 through the analytical lens of protein domain architecture and structural organization, a core tenet of our broader thesis research. The precise arrangement of Cas9's functional domains—nucleases, recognition lobes, and linker regions—directly dictates its mechanistic action, specificity, and engineerability.
Evolutionary Origin: Bacterial Adaptive Immunity
In bacteria and archaea, CRISPR-Cas systems provide acquired immunity against invading phages and plasmids. The process involves three stages:
1. Adaptation: Cas1-Cas2 complexes capture short fragments of foreign DNA (protospacers) and integrate them into the host's CRISPR array as new spacers. 2. Expression: The CRISPR array is transcribed and processed into short CRISPR RNAs (crRNAs). 3. Interference: A crRNA guides the Cas effector complex (e.g., Cas9) to complementary foreign DNA, leading to its cleavage and degradation.
A key feature of Type II systems, which include Cas9, is the requirement of a protospacer adjacent motif (PAM) in the target DNA, a critical specificity determinant encoded in the protein's PAM-interacting domain.
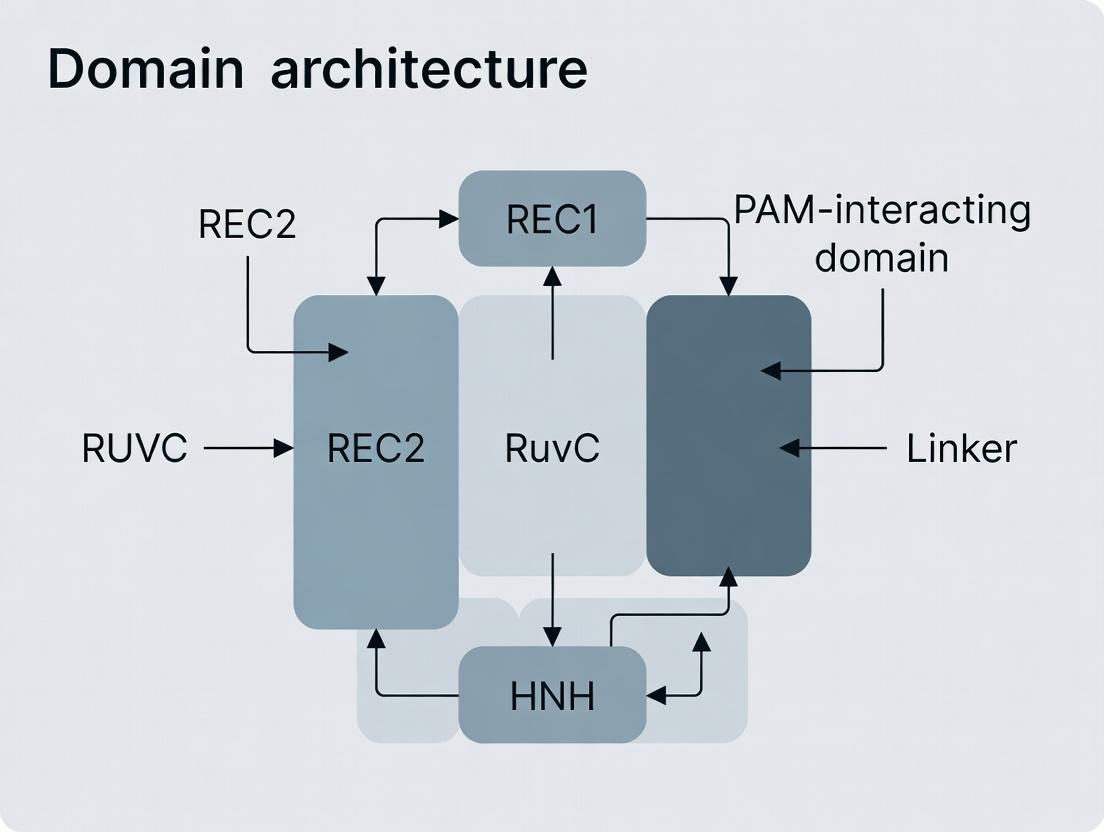
Cas9 Protein Domain Architecture and Mechanism
Streptococcus pyogenes Cas9 (SpCas9) is the archetypal and most widely engineered variant. Its structure is organized into distinct lobes and domains that coordinate nucleic acid binding and cleavage.
Table 1: Core Structural Domains of S. pyogenes Cas9 (SpCas9)
| Domain/Lobe | Amino Acid Residues (Approx.) | Primary Function | Architectural Role |
|---|---|---|---|
| REC Lobe (Recognition) | 1-180, 310-713 | Facilitates sgRNA and target DNA binding, allosteric regulation | Provides the structural scaffold for nucleic acid hybridization monitoring. |
| Bridge Helix | 60-93 | Unwinds DNA duplex during R-loop formation | Acts as a flexible molecular hinge between lobes. |
| REC1 & REC2 | - | Direct sgRNA:DNA heteroduplex interaction | Critical for target DNA melting and specificity. |
| NUC Lobe (Nuclease) | 181-309, 714-1368 | Contains nuclease activity and PAM recognition | Executes the catalytic function; houses the PAM sensor. |
| PAM-Interacting (PI) Domain | 1099-1368 | Reads the 5'-NGG-3' PAM sequence | Key determinant of target site specificity and discrimination. |
| HNH Nuclease Domain | 775-908 | Cleaves the DNA strand complementary to the crRNA (target strand) | Positioned within the catalytic core; requires structural activation. |
| RuvC-like Nuclease Domain | 1-59, 718-775, 909-1098 | Cleaves the non-complementary DNA strand (non-target strand) | Composed of three split subdomains; structurally analogous to retroviral integrases. |
The mechanism involves:
- PAM Recognition: The PI domain scans DNA for an NGG PAM, initiating binding.
- DNA Melting: PAM binding induces local DNA unwinding, facilitated by the REC lobe.
- R-loop Formation: The crRNA guide strand hybridizes with the target DNA strand, displacing the non-target strand.
- Conformational Activation: Successful heteroduplex formation triggers a allosteric signal from the REC lobe to the NUC lobe.
- Double-Strand Break (DSB) Cleavage: The HNH domain cleaves the target strand; the RuvC domain cleaves the non-target strand, generating a blunt-ended DSB 3 bp upstream of the PAM.
Diagram 1: Cas9 DNA Targeting and Cleavage Cascade
Key Experimental Protocols for Cas9 Function Analysis
Protocol 1:In VitroDNA Cleavage Assay (PAM Validation)
Purpose: To verify Cas9 nuclease activity and define PAM requirements. Materials: Purified Cas9 protein, in vitro transcribed sgRNA, linear dsDNA substrate with candidate PAM sequences. Procedure:
- Assembly: Combine 100 nM Cas9, 120 nM sgRNA, and 10 nM target DNA in nuclease buffer (20 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl₂, 5% glycerol).
- Incubation: React at 37°C for 60 minutes.
- Termination: Add Proteinase K and EDTA to final concentrations of 0.2 mg/mL and 10 mM, respectively. Incubate at 56°C for 15 min.
- Analysis: Resolve products on a 2% agarose gel stained with ethidium bromide. Cleavage efficiency is quantified as the fraction of linear substrate converted to shorter fragments.
Protocol 2: Cellular Genome Editing & Indel Analysis (T7E1 Assay)
Purpose: To measure Cas9-mediated indel (insertion/deletion) formation at an endogenous genomic locus in mammalian cells. Procedure:
- Transfection: Deliver a Cas9 expression plasmid and a sgRNA expression construct into HEK293T cells (e.g., via lipofection).
- Harvest: 72 hours post-transfection, extract genomic DNA using a silica-column kit.
- PCR Amplification: Amplify the target genomic region (200-400 bp) using high-fidelity polymerase.
- Heteroduplex Formation: Denature and reanneal PCR products (95°C for 10 min, ramp to 85°C at -2°C/s, then to 25°C at -0.1°C/s).
- Nuclease Digestion: Treat with T7 Endonuclease I (T7E1), which cleaves mismatched heteroduplex DNA formed by wild-type and mutant alleles.
- Quantification: Analyze fragments by agarose gel electrophoresis. Indel frequency (%) is calculated using the formula: 100 × (1 - sqrt(1 - (b+c)/(a+b+c))), where a is the integrated intensity of undigested product, and b+c are the digested fragment intensities.
Diagram 2: T7E1 Assay for Genome Editing Efficiency
Quantitative Data on Cas9 Variants and Performance
Table 2: Comparative Analysis of Engineered Cas9 Variants
| Cas9 Variant | Parent | Key Modifications | Average On-Target Efficiency | Reported Off-Target Reduction | Primary Application |
|---|---|---|---|---|---|
| Wild-type SpCas9 | S. pyogenes | N/A | 40-70% (varies by locus/cell) | Baseline | General DSB generation |
| SpCas9-HF1 | SpCas9 | N497A/R661A/Q695A/Q926A (weaken DNA binding) | Comparable to WT | ~10-fold reduction | High-fidelity editing |
| eSpCas9(1.1) | SpCas9 | K848A/K1003A/R1060A (alter electrostatic interactions) | Comparable to WT | ~10-fold reduction | High-fidelity editing |
| xCas9 | SpCas9 | A262T/R324L/S409I/E480K/E543D/M694I/E1219V (directed evolution) | Broad PAM (NG, GAA, GAT), efficiency varies | Up to 1,400-fold reduction | Expanded targeting range |
| SpCas9-NG | SpCas9 | R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R | Recognizes NG PAM, ~50-70% of NGG efficiency | Comparable to WT | Expanded NG PAM targeting |
| SaCas9 | S. aureus | Ortholog, smaller size | 10-50% (lower than SpCas9) | Generally lower than SpCas9 | In vivo delivery (AAV compatible) |
Table 3: Common DSB Repair Outcome Frequencies in Mammalian Cells
| Repair Pathway | Typical Timeframe | Dominant Outcome | Relative Frequency | Experimental Modulation |
|---|---|---|---|---|
| Non-Homologous End Joining (NHEJ) | Minutes to Hours | Small insertions/deletions (Indels) | ~60-80% (error-prone) | Inhibited by DNA-PKcs inhibitors (e.g., NU7026) |
| Microhomology-Mediated End Joining (MMEJ) | ~1 Hour | Deletions flanked by microhomology | ~10-20% | Inhibited by Polθ inhibition |
| Homology-Directed Repair (HDR) | Hours to Days | Precise edits (with donor template) | Typically <10% (varies with cell cycle) | Enhanced by synchronizing cells in S/G2 phase; inhibited by NHEJ inhibitors. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Cas9-Based Genome Editing Research
| Reagent / Material | Supplier Examples | Function & Critical Notes |
|---|---|---|
| Recombinant S. pyogenes Cas9 Nuclease | NEB, Thermo Fisher, IDT | High-purity, ready-to-use protein for in vitro assays (cleavage, RNP delivery). |
| Custom sgRNA (synthetic, crRNA:tracrRNA, or plasmid) | IDT, Synthego, Sigma-Aldrich | Provides targeting specificity. Chemical modifications can enhance stability. |
| T7 Endonuclease I (T7E1) | NEB | Mismatch-specific nuclease for rapid quantification of indel formation. |
| Surveyor / Cel-I Nuclease | IDT | Alternative mismatch-specific nuclease for indel detection. |
| High-Fidelity DNA Polymerase (for amplicon sequencing) | NEB (Q5), Takara (PrimeSTAR) | Essential for error-free amplification of target loci prior to sequencing or T7E1 assay. |
| Next-Generation Sequencing Library Prep Kit | Illumina, Twist Bioscience | For deep sequencing (e.g., amplicon-seq) to comprehensively profile editing outcomes and off-targets. |
| Lipofectamine CRISPRMAX Transfection Reagent | Thermo Fisher | Optimized lipid nanoparticle for delivering Cas9 RNP or plasmid DNA into hard-to-transfect cells. |
| AAV Packaging System (for in vivo delivery) | Addgene (plasmids), Vigene Biosciences | Required for packaging SaCas9 or smaller Cas9 variants into AAV vectors for animal studies. |
| Anti-Cas9 Monoclonal Antibody | Abcam, Cell Signaling Tech | For Western blot, ELISA, or immunoprecipitation to verify Cas9 expression or cellular localization. |
| Guide-it CRISPR Validation Kit | Takara Bio | Integrated solution for T7E1-based screening of sgRNA activity. |
The transformative power of Cas9 as a molecular scissor is a direct consequence of its modular protein architecture. Our structural organization thesis underscores that each domain—from the REC lobe's role in fidelity to the split RuvC domain's catalytic mechanism—represents a discrete unit for rational engineering. Advances like high-fidelity (HF-Cas9) and PAM-relaxed (xCas9, SpCas9-NG) variants exemplify how atomic-level structural insights drive functional optimization. Future drug development and therapeutic genome editing will continue to rely on deconstructing and reconfiguring this elegant molecular machine to achieve unprecedented precision and control.
Within the broader thesis on Cas9 protein domain architecture, the bilobed organization into Recognition (REC) and Nuclease (NUC) lobes represents a fundamental structural paradigm essential for target DNA interrogation and cleavage. This whitepaper provides an in-depth technical analysis of this architecture, its functional consequences, and methodologies for its study, serving as a critical resource for therapeutic development.
Structural Anatomy of the Bilobed Architecture
Cas9 undergoes a large conformational rearrangement upon guide RNA binding, forming the characteristic bilobed structure. The lobes are connected by a linker helix.
Recognition Lobe (REC): Primarily α-helical, responsible for guide RNA and target DNA strand recognition and binding fidelity.
- Key Subdomains: REC1, REC2, REC3. REC1 interacts with the repeat:anti-repeat duplex of the guide RNA.
- Function: Governs sgRNA loading, DNA target searching, and sequence specificity. Mutations here often affect cleavage fidelity.
Nuclease Lobe (NUC): Contains the conserved HNH and RuvC-like nuclease domains, along with the PAM-interacting (PI) domain.
- HNH Domain: Cleaves the complementary (target) DNA strand.
- RuvC Domain: Cleaves the non-complementary (non-target) DNA strand.
- PI Domain: Directly reads the Protospacer Adjacent Motif (PAM), triggering local DNA unwinding.
Interface and Cleavage Cavity: The cleft between the REC and NUC lobes forms a positively charged channel where DNA binding and catalysis occur.
Table 1: Quantitative Comparison of S. pyogenes Cas9 (SpCas9) Lobes
| Parameter | REC Lobe | NUC Lobe | Notes |
|---|---|---|---|
| Approx. Residue Range | 1-59, 718-775, 909-1098 | 60-717, 776-908 | UniProt P99ZF4 |
| Molecular Weight (kDa) | ~45 kDa | ~95 kDa | Full-length SpCas9 ~160 kDa |
| Key Structural Motifs | Helical Bundle, Bridge Helix | HNH, RuvC (ββα-metal folds), PI (β-sheet) | |
| Key Functional Residues | R66, K455, K526 (DNA binding) | D10, H840 (Catalytic), R1333/R1335 (PAM read) | Mutations D10A/H840A create "dCas9" |
| % of Mutations Affecting Fidelity | ~65% | ~35% | Based on deep mutational scanning data |
Experimental Protocols for Studying Lobe Dynamics
Protocol: Single-Molecule FRET to Monitor Lobe Conformational Changes
Objective: Measure real-time dynamics of REC-NUC lobe opening/closing during DNA engagement.
Methodology:
- Labeling: Site-specifically label Cas9 with donor (Cy3) on the REC lobe (e.g., residue A90C) and acceptor (Cy5) on the NUC lobe (e.g., residue S355C) using maleimide chemistry.
- Immobilization: Biotinylate Cas9 at the C-terminus and tether it to a PEG-passivated, streptavidin-coated quartz microscope slide.
- Imaging: Use a total-internal-reflection fluorescence (TIRF) microscope. Illuminate with 532 nm laser to excite donor.
- Data Acquisition:
- Flow in buffer containing 100 nM sgRNA and 50 nM target DNA duplex.
- Record donor and acceptor emission intensities over time (≥ 30 fps).
- Calculate FRET efficiency (EFRET) = IA / (ID + IA), where I= intensity.
- Analysis: Plot EFRET vs. time. High FRET indicates closed, active state; low FRET indicates open, inactive state. Perform hidden Markov modeling to derive transition kinetics.
Protocol: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
Objective: Map allosteric communication and surface accessibility changes between lobes upon ligand binding.
Methodology:
- Sample Preparation: Prepare four states: apo-Cas9, Cas9:sgRNA, Cas9:sgRNA:target DNA, Cas9:sgRNA:non-target DNA.
- Deuterium Labeling: Dilute protein complex 1:10 into D2O buffer (pD 7.0, 25°C). Quench at time points (3s, 30s, 300s, 3000s) with cold, low-pH quench buffer (final pH 2.5).
- Digestion & Analysis: Pass quenched sample over immobilized pepsin column. Analyze peptides by LC-MS. Monitor mass shift due to deuterium uptake.
- Data Processing: Calculate differential HDX (ΔHDX) between states. Regions with decreased ΔHDX upon binding indicate protected interfaces or allosteric changes.
Visualization of Functional Dynamics
Diagram 1: Cas9 activation pathway from apo to cleaving state.
Diagram 2: Functional division of labor between REC and NUC lobes.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Bilobed Architecture Studies
| Reagent | Function & Application | Example Product/Source |
|---|---|---|
| Site-Specific Labeling Kits (e.g., SNAP-, HALO-, CLIP-tag) | For covalent, specific attachment of fluorophores (FRET pairs) or biotin to engineered tags on specific lobes. | New England Biolabs SNAP-Surface dyes |
| Biotinylated Cas9 Variants | For surface immobilization in single-molecule or binding assays (SPR, BLI). | Thermo Fisher Scientific, custom from IDT |
| HDX-MS Software Suites | For automated peptide analysis, deuterium uptake calculation, and visualization (e.g., HDExaminer). | Sierra Analytics HDExaminer |
| Stable Isotope-Labeled Proteins (¹⁵N, ¹³C) | For NMR studies of lobe dynamics and allostery. | Produced in E. coli using labeled media (Cambridge Isotopes) |
| Cryo-EM Grids & Vitrobots (e.g., Quantifoil, UltrAuFoil) | For high-resolution structural analysis of multiple conformational states. | EMS Diasum |
| Dual-Luciferase Reporter Assay Systems | For high-throughput functional screening of Cas9 lobe mutants for fidelity/activity. | Promega |
| Mobility Shift Assay (EMSA) Kits | To qualitatively assess DNA binding competency of lobe mutants. | Thermo Fisher Scientific LightShift Chemiluminescent EMSA Kit |
| Surface Plasmon Resonance (SPR) Chips (e.g., NTA, CM5) | For kinetic analysis of lobe-dependent protein-DNA/RNA interactions. | Cytiva Series S Sensor Chips |
Within the broader thesis on Cas9 protein domain architecture, the nuclease (NUC) lobe is the catalytic heart responsible for programmable DNA cleavage. This lobe comprises two distinct nuclease domains: HNH and RuvC. The HNH domain cleaves the complementary (target) DNA strand, while the RuvC domain cleaves the non-complementary (non-target) strand. This in-depth guide explores the structural organization, cleavage mechanisms, and experimental characterization of these critical domains.
Domain Structure and Catalytic Motifs
The HNH domain is a ββα-metal fold domain that inserts into the major groove of the DNA:RNA heteroduplex. It contains a catalytic metal-binding site, typically coordinated by conserved histidine and asparagine residues. The RuvC domain, homologous to the RNase H superfamily, adopts a retroviral integrase-like fold and contains a catalytic triad of acidic residues (D10, E762, H983 in S. pyogenes Cas9) that coordinate Mg²⁺ ions for hydrolysis.
Table 1: Key Structural Features of Cas9 Nuclease Domains
| Feature | HNH Domain | RuvC Domain |
|---|---|---|
| Structural Fold | ββα-metal fold | RNase H/Retroviral integrase fold |
| Catalytic Motif | HNH motif (e.g., H840, N854, H858 in SpCas9) | DEDH motif (e.g., D10, E762, D855, H983 in SpCas9) |
| Metal Ion Cofactor | Mg²⁺ (primary) | Two Mg²⁺ ions (in a two-metal-ion mechanism) |
| DNA Strand Targeted | Complementary (Target) Strand | Non-complementary (Non-target) Strand |
| Cleavage Position | 3 bp upstream of PAM | 3 bp upstream of PAM on opposite strand |
Cleavage Mechanisms and Activation
Cleavage is a multi-step, conformationally gated process. Upon correct target DNA recognition and R-loop formation, the HNH domain undergoes a large-scale (~35 Å) conformational rotation to engage the target strand. The RuvC domain remains relatively static but its active site becomes accessible only upon displacement of the non-target strand.
Table 2: Quantitative Kinetics of Cas9 Cleavage (Representative Data)
| Parameter | HNH Domain Cleavage | RuvC Domain Cleavage | Experimental Method |
|---|---|---|---|
| Catalytic Rate (kcat) | ~0.5 – 2.0 min⁻¹ | ~0.1 – 1.0 min⁻¹ | Single-turnover kinetics (stopped-flow) |
| Metal Ion Dependence (Km) | [Mg²⁺] ~ 1-2 mM | [Mg²⁺] ~ 0.5-1 mM | Metal titration with fluorescent DNA substrates |
| Cleavage Timing | Can precede or be synchronous with RuvC | Often follows HNH activation | Quenched-flow, time-resolved crystallography |
Diagram 1: Conformational Activation of Cas9 Nuclease Domains
Experimental Protocols for Nuclease Analysis
Protocol 1: In Vitro Cleavage Assay with Fluorescently-Labeled DNA Substrates Objective: Determine cleavage efficiency and kinetics of HNH vs. RuvC activity.
- Substrate Preparation: Synthesize DNA duplexes containing the target sequence and PAM. Label the 5’ end of the target strand with a fluorophore (e.g., FAM) and the non-target strand with a different fluorophore (e.g., Cy5). Use a quencher on the 3’ end for real-time assays.
- Reaction Setup: In a 20 µL reaction, combine 50 nM Cas9:sgRNA complex (pre-assembled), 10 nM labeled DNA substrate, in buffer (20 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl₂, 1 mM DTT, 5% glycerol). MgCl₂ is added last to initiate reaction.
- Kinetic Measurement: Aliquot reactions into a 96-well plate. Monitor fluorescence de-quenching (ex/em: 492/518 nm for FAM; 649/670 nm for Cy5) in a real-time PCR instrument or plate reader at 37°C for 30-60 minutes.
- Data Analysis: Calculate initial velocities. Use polyacrylamide gel electrophoresis (PAGE) with urea for endpoint analysis to separate cleaved and uncleaved products, visualized by a fluorescence gel scanner.
Protocol 2: Single-Molecule FRET (smFRET) to Probe Domain Conformation Objective: Observe real-time conformational dynamics of the HNH domain.
- Dye Labeling: Introduce cysteine mutations at specific sites on the HNH domain (e.g., S867C) and the REC lobe (e.g., S355C). Purify and label with maleimide-conjugated donor (Cy3) and acceptor (Cy5) dyes.
- Surface Immobilization: Passivate a quartz microfluidic chamber with PEG-biotin. Introduce streptavidin, then biotinylated DNA substrates.
- Data Acquisition: Dilute labeled Cas9:sgRNA to ~50 pM and flow into chamber. Image using a total-internal-reflection fluorescence (TIRF) microscope. Monitor donor and acceptor emission simultaneously upon 532 nm laser excitation.
- Analysis: Calculate FRET efficiency (E = IA/(ID+IA)) for individual molecules over time. Identify transitions between low-FRET (HNH swung away) and high-FRET (HNH engaged) states correlated with DNA binding.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Nuclease Lobe Research
| Reagent/Material | Function/Description | Example Supplier/Product |
|---|---|---|
| Wild-type & Catalytic Mutant Cas9 (D10A, H840A) | Control proteins for dissecting individual domain activity; D10A (RuvC- dead), H840A (HNH-dead). | Purified from E. coli expression systems or commercial vendors (e.g., NEB, Thermo Fisher). |
| Fluorophore/Quencher-labeled DNA Oligos | Substrates for real-time, continuous cleavage assays and strand-specific activity measurement. | Custom synthesis from IDT or Eurofins with modifications like 5’-FAM/3’-Iowa Black FQ. |
| High-Purity MgCl₂ & Metal Chelators (EDTA, EGTA) | Essential cofactor manipulation; chelators used to initiate/stop reactions and probe metal dependence. | Molecular biology grade, Sigma-Aldrich. |
| Single-Cysteine Mutant Cas9 Proteins | Site-specific labeling for smFRET, EPR, or other biophysical conformational studies. | Generated via site-directed mutagenesis kits (e.g., Q5 from NEB). |
| Stopped-Flow or Quenched-Flow Apparatus | For measuring rapid cleavage kinetics in the millisecond to second timescale. | Instruments from Applied Photophysics or KinTek. |
| Anti-Cas9 Monoclonal Antibodies (Domain-Specific) | For immunoprecipitation, Western blot, or inhibiting specific domains in cellular assays. | Available from Abcam, Cell Signaling Technology. |
Diagram 2: Workflow for Strand-Specific Cleavage Kinetics Assay
Understanding the precise structure and mechanism of the NUC lobe’s HNH and RuvC domains is foundational for CRISPR-Cas9 engineering. This knowledge directly enables the development of high-fidelity variants, nickases, and entirely novel editors (e.g., base editors that exploit a disabled RuvC domain). For drug development, targeting these domains with small-molecule inhibitors offers a potential strategy for controlling CRISPR activity in therapeutic contexts, underscoring the critical role of fundamental domain architecture research in applied biotechnology.
Within the structural architecture of the CRISPR-Cas9 enzyme, the Recognition (REC) lobe is a critical catalytic domain responsible for orchestrating key steps in target DNA interrogation. This whitepaper, framed within a broader thesis on Cas9 protein domain architecture and structural organization, details the mechanistic role of the REC lobe. Comprising the REC1, REC2, and REC3 subdomains, this lobe facilitates sgRNA stabilization, mediates the DNA melting bubble formation, and participates in heteroduplex formation and specificity verification. Its conformational dynamics are integral to the transition from a DNA surveillance state to an active cleavage complex.
Structural Organization of the REC Lobe
The REC lobe is a predominantly α-helical domain that bridges the nucleic acid-binding channel and the nuclease (NUC) lobe containing RuvC and HNH domains.
Table 1: REC Lobe Subdomains and Primary Functions
| Subdomain | Structural Features | Primary Role in Cas9 Function |
|---|---|---|
| REC1 | Large, central helical domain | Major contributor to sgRNA binding; mediates conformational activation upon PAM recognition. |
| REC2 | Bridge helix and adjacent loops | Critical for stabilizing the nontarget DNA strand; involved in initial DNA melting. |
| REC3 | Smaller, variable region | Contributes to target strand positioning and discrimination against mismatches near the PAM. |
Functional Mechanisms
sgRNA Binding and Complex Assembly
The REC lobe, particularly REC1, forms extensive contacts with the repeat:anti-repeat duplex of the sgRNA. This binding is essential for maintaining the ribonucleoprotein (RNP) complex in a conformationally poised state prior to DNA encounter. Structural studies indicate that REC lobe interactions pre-organize the guide RNA seed region for optimal base pairing with target DNA.
DNA Melting and R-Loop Formation
Upon PAM recognition by the C-terminal domain of the NUC lobe, a signal is transduced to the REC lobe, triggering large-scale conformational changes. The REC2 and REC3 domains facilitate the unwinding (melting) of the double-stranded DNA. The REC lobe, specifically the bridge helix within REC2, acts as a wedge to separate DNA strands, enabling the formation of the RNA-DNA heteroduplex (R-loop).
Table 2: Quantitative Parameters of REC-Lobe Mediated DNA Melting (Streptococcus pyogenes Cas9)
| Parameter | Value/Measurement | Experimental Method |
|---|---|---|
| Energetic Contribution to DNA Unwinding | ~ -8.6 kcal/mol (estimated) | Single-molecule FRET, Thermodynamic modeling |
| Conformational Shift upon PAM Binding | ~ 10-15 Å movement of REC lobes | Cryo-EM, X-ray Crystallography |
| Rate of R-loop Propagation (5' to 3') | ~ 10-30 base pairs/second | Single-molecule Magnetic Tweezers |
| Impact of REC3 Deletion on Cleavage Efficiency | Reduction to 1-5% of wild-type activity | In vitro Cleavage Assay |
Target Recognition and Specificity
The REC lobe is a major determinant of Cas9's specificity. REC3 acts as a "mismatch sensor" for bases proximal to the PAM. Mismatches in this region induce structural distortions that are amplified by the REC lobe, leading to inhibition of HNH nuclease domain activation and aborting the cleavage pathway. This provides a critical proofreading step to minimize off-target effects.
Experimental Protocols for Investigating REC Lobe Function
Protocol: Site-Directed Mutagenesis andIn VitroCleavage Assay
Purpose: To assess the functional impact of specific residues in REC subdomains.
- Design: Identify target residues in REC1/REC2/REC3 via sequence alignment and structural data (PDB: 4UN3).
- Mutagenesis: Perform PCR-based site-directed mutagenesis on the Cas9 expression plasmid (e.g., pET-based). Verify by Sanger sequencing.
- Protein Purification: Express mutant and wild-type His6-tagged Cas9 in E. coli BL21(DE3). Purify via Ni-NTA affinity chromatography, followed by size-exclusion chromatography (Superdex 200).
- In Vitro Transcription: Generate target sgRNA using T7 RNA polymerase.
- Cleavage Reaction: Assemble RNP complex (100 nM Cas9, 120 nM sgRNA) in reaction buffer (20 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT). Incubate 10 min at 25°C. Add linearized target DNA plasmid (10 nM). Incubate 30-60 min at 37°C.
- Analysis: Resolve products on 1% agarose gel. Quantify cleavage efficiency via gel densitometry.
Protocol: Single-Molecule FRET (smFRET) to Monitor REC Conformation
Purpose: To measure real-time conformational dynamics of the REC lobe during DNA engagement.
- Labeling: Engineer single cysteine mutations in the REC lobe (e.g., REC1) and a reference point on the NUC lobe. Label with maleimide-conjugated donor (Cy3) and acceptor (Cy5) fluorophores.
- Surface Immobilization: Biotinylate a dual-labeled Cas9 protein and immobilize on a streptavidin-coated quartz microscope slide in imaging buffer with oxygen scavengers.
- Flow Cell Experiment: Introduce sgRNA, followed by target DNA containing/ lacking a PAM or with mismatches.
- Data Acquisition: Image using a TIRF microscope. Track FRET efficiency (EFRET) over time for hundreds of individual molecules.
- Analysis: Generate EFRET histograms and transition density plots to identify conformational states and their lifetimes.
Protocol: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)
Purpose: To map regions of the REC lobe involved in sgRNA/DNA binding and to identify allosteric changes.
- Sample Preparation: Incubate apo-Cas9, Cas9:sgRNA, and Cas9:sgRNA:target DNA complexes in deuterated buffer (e.g., 99.9% D2O, pD 7.5) for varying time points (10s to 2 hours).
- Quenching & Digestion: Quench exchange by lowering pH and temperature. Digest with immobilized pepsin.
- LC-MS/MS Analysis: Inject peptides onto a UPLC system at 0°C, followed by ESI-TOF mass spectrometry.
- Data Processing: Identify peptides and calculate deuterium uptake for each time point. Differences in uptake >5% between states are considered significant, highlighting regions (e.g., REC2 loops) involved in binding or conformational change.
Visualization of REC Lobe Mechanisms
Title: REC Lobe Role in Cas9 Activation and Target Verification Pathway
The Scientist's Toolkit: Key Research Reagents & Materials
Table 3: Essential Research Reagents for Investigating the REC Lobe
| Item | Function/Application | Example (Supplier) |
|---|---|---|
| Recombinant Wild-Type & Mutant Cas9 Proteins | Substrate for structural, biochemical, and biophysical assays. Critical for studying REC domain mutations. | SpyCas9 (NEB, Thermo Fisher) |
| Synthetic sgRNAs (Chemically Modified) | For forming defined RNP complexes. 2'-O-methyl and phosphorothioate modifications enhance stability for in vitro assays. | Synthesized via IDT or Trilink. |
| Fluorescent Nucleotide/Dye Conjugates | For labeling DNA substrates (smFRET, EMSA) or protein (cysteine/maleimide chemistry) to monitor binding and dynamics. | Cy3/Cy5 maleimide (Lumiprobe), ATTO dyes (Sigma). |
| HDX-MS Buffer & Quenching Solutions | Specialized buffers for deuterium exchange experiments, including low pH, low temperature quench to preserve exchange state. | HDX-MS Buffer Kit (Waters Corp). |
| Size-Exclusion Chromatography Columns | For purifying monodisperse, stable Cas9 protein and protein-nucleic acid complexes for structural work. | Superdex 200 Increase (Cytiva). |
| Cryo-EM Grids & Vitrification System | For high-resolution structural determination of Cas9-REC lobe conformations in different functional states. | Quantifoil grids, Vitrobot (Thermo Fisher). |
| Single-Molecule Imaging Flow Cells | Customizable chambers for TIRF microscopy-based smFRET and tethered particle motion assays. | Streptavidin-coated microfluidic cells (Microsurfaces Inc.). |
| PAM-Disabled or Mismatch-Containing DNA Libraries | To probe the specificity contribution of the REC lobe in high-throughput in vitro or cellular assays. | Custom array-synthesized oligo pools (Twist Bioscience). |
The REC lobe is the central processing unit of the Cas9 enzyme, integrating sgRNA binding, PAM-induced signals, and mismatch detection to govern DNA cleavage fidelity. Its architecture and dynamics are fundamental to understanding CRISPR-Cas9 function. Ongoing research into REC lobe engineering aims to modulate its allosteric control, creating high-fidelity and hyper-accurate Cas9 variants with critical applications in therapeutic genome editing and diagnostic technologies. This exploration forms a cornerstone of the comprehensive thesis on Cas9 domain architecture, highlighting how individual lobes synergize to execute precise genome surgery.
Within the broader thesis investigating Cas9 protein domain architecture and structural organization, this whitepaper provides an in-depth technical analysis of three critical functional modules: the PAM-Interacting Domain (PID), the inter-domain linkers, and the helical bridge motifs. These elements collectively govern DNA target recognition, allosteric signal transduction, and structural integrity, making them pivotal for understanding Cas9 mechanics and for therapeutic engineering.
The CRISPR-Cas9 system's precision stems from its multi-domain architecture. Beyond the well-characterized RuvC and HNH nuclease lobes, the PID, flexible linkers, and helical bridges serve as essential regulatory and structural components. This guide dissects their roles within the holistic Cas9 structural framework, providing a foundation for rational protein engineering aimed at enhancing specificity, altering PAM requirements, and developing novel gene-editing tools.
PAM-Interacting Domain (PID)
Structural and Functional Role
The PID, often located in the C-terminal region of Cas9 (e.g., in Streptococcus pyogenes Cas9), is responsible for initiating target DNA binding by recognizing a short Protospacer Adjacent Motif (PAM). This recognition triggers local DNA melting and facilitates subsequent R-loop propagation.
Key Quantitative Data (SpyCas9):
Table 1: PAM-Interacting Domain Characteristics for SpyCas9
| Property | Value / Description | Experimental Method |
|---|---|---|
| Primary Location | C-terminal domain (CTD) | X-ray crystallography, Cryo-EM |
| Canonical PAM Sequence | 5'-NGG-3' | In vitro cleavage assays, SELEX |
| Critical Recognition Residues | R1333, R1335, T1337, Y1349 | Alanine-scanning mutagenesis |
| Binding Affinity (to PAM DNA) | Kd ~ 30-100 nM | Surface Plasmon Resonance (SPR) |
| Effect on Catalytic Rate | PAM binding increases kcat by ~1000-fold | Stopped-flow kinetics |
Experimental Protocol: Assessing PAM Specificity via High-Throughput Sequencing
Objective: To quantitatively determine the PAM preferences for a wild-type or engineered Cas9 variant. Methodology:
- Library Construction: Synthesize a degenerate oligonucleotide library containing a randomized PAM region (e.g., NNNN) flanking a constant protospacer sequence.
- In Vitro Cleavage: Incubate the purified Cas9 protein complexed with sgRNA with the DNA library in appropriate buffer. Halt the reaction after partial digestion.
- Selection of Cleaved Products: Use gel electrophoresis or size selection beads to isolate the cleaved DNA fragments.
- Sequencing & Analysis: Amplify the selected fragments via PCR and subject them to high-throughput sequencing. Align reads to the reference library and compute the enrichment or depletion of each PAM sequence in the cleaved pool versus the initial library to generate a PAM logo.
Inter-Domain Linkers
Role in Allostery and Dynamics
Linkers are not merely passive connectors; they act as flexible hinges and allosteric regulators. Their length and composition influence the large-scale conformational transitions between the catalytically inactive "apo" state and the active DNA-bound state.
Table 2: Characteristics of Major Cas9 Linkers
| Linker Name/Region | Connects | Role in Mechanism | Key Mutagenesis Findings |
|---|---|---|---|
| L1/L2 (Bridge Helix) | RuvC & Rec lobes | Nucleotide flipping, catalysis | Rigidifying mutations reduce cleavage efficiency. |
| HNH-Domain Linker | HNH nuclease & Rec lobe | Positions HNH for cleavage | Shortening linker decouples HNH activation. |
| RuvC-Connecter Linker | RuvC nuclease & CTD (PID) | Transmits PAM signal to RuvC | Glycine insertion increases off-target activity. |
Helical Bridges
Structural Stabilization and Signal Relay
Helical bridges are conserved alpha-helical bundles that act as central scaffolds, holding major lobes together. They are critical for transmitting the conformational change initiated by PAM binding at the PID to the distant nuclease active sites.
Experimental Protocol: Probing Conformational Dynamics via smFRET
Objective: To measure real-time conformational changes in linkers and helical bridges upon DNA binding. Methodology:
- Sample Labeling: Engineer cysteine residues at specific positions within a linker or on either side of a helical bridge in a cysteine-null Cas9 background. Label these sites with appropriate donor (e.g., Cy3) and acceptor (Cy5) fluorophores.
- Imaging: Immobilize labeled Cas9:sgRNA complexes on a passivated microscope slide.
- Data Acquisition: Use a total-internal-reflection fluorescence (TIRF) microscope to excite the donor and record emission intensities of both donor and acceptor over time for individual molecules.
- Triggering & Analysis: Introduce target DNA into the flow chamber. Calculate FRET efficiency (E = IA/(ID+IA)) over time. Observe shifts in FRET states, which correspond to discrete conformational changes (e.g., opening/closing of lobes, HNH movement).
Integrated View: Signaling Pathway from Recognition to Cleavage
Diagram Title: Cas9 Activation Pathway from PAM Binding to DNA Cleavage
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Cas9 Domain Architecture Studies
| Reagent / Material | Supplier Examples | Function in Research |
|---|---|---|
| Site-Directed Mutagenesis Kits | NEB Q5, Agilent QuikChange | Introducing point mutations in PID, linkers, or bridges. |
| Fluorophore Dyes (Cy3, Cy5, Alexa Fluor) | Lumiprobe, Thermo Fisher | Labeling engineered cysteines for smFRET dynamics studies. |
| Streptavidin-Coated Slides/Chambers | Microsurfaces, Ibidi | Immobilizing biotinylated Cas9/sgRNA for single-molecule imaging. |
| Gel Filtration/SEC Columns | Cytiva, Bio-Rad | Purifying Cas9 protein complexes for structural studies. |
| Degenerate Oligo PAM Libraries | IDT, Twist Bioscience | Profiling PAM specificity for wild-type and engineered PIDs. |
| Anti-Cas9 Monoclonal Antibodies | Diagenode, Abcam | Immunoprecipitation (IP) for pull-down assays of domain mutants. |
| HPLC-Purified sgRNA | Synthego, Trilink | Ensuring consistent ribonucleoprotein complex formation. |
The PAM-Interacting Domain, inter-domain linkers, and helical bridges constitute the core regulatory and mechanical infrastructure of Cas9. A detailed understanding of their synergistic function within the overall protein architecture is indispensable. This knowledge directly enables the rational design of next-generation editors with altered PAMs, reduced off-target effects, and novel functionalities, thereby advancing therapeutic genome engineering.
1. Introduction: Structural Insights into CRISPR-Cas9 Function
The precision of CRISPR-Cas9 genome editing is a direct consequence of its programmable, multi-component architecture. A comprehensive understanding of Cas9 protein domain organization and its orchestrated assembly with the sgRNA and target DNA into a catalytically active ternary complex is foundational for ongoing research. This whitepaper, framed within a broader thesis on Cas9 structural biology, provides an in-depth technical guide to the assembly mechanics and visualization of this critical complex, which directly informs the engineering of next-generation editors and therapeutic agents.
2. Structural Architecture and Assembly Dynamics
The ternary complex formation is a multi-step process involving significant conformational rearrangements. The core domains of Streptococcus pyogenes Cas9 (SpCas9) and their roles are detailed below.
Table 1: Key Domains of SpCas9 and Their Functions in Ternary Complex Assembly
| Domain/Acceptor | Primary Function in Assembly | Key Structural Outcome |
|---|---|---|
| REC Lobe (Recognition Lobe) | Facilitates sgRNA and DNA binding; undergoes major conformation change. | Positions the sgRNA:DNA heteroduplex for cleavage; critical for PAM recognition. |
| REC I, II, III | ||
| NUC Lobe (Nuclease Lobe) | Contains the two catalytic centers and the PAM-interaction site. | Executes DNA cleavage upon successful heteroduplex formation. |
| HNH Domain | Cleaves the target DNA strand complementary to the sgRNA. | Rotates into position upon strand invasion. |
| RuvC Domain | Cleaves the non-target DNA strand. | Active site is pre-formed; cleaves post-HNH activation. |
| PI (PAM-Interacting) Domain | Reads the 5'-NGG-3' PAM sequence in the target DNA. | Initiates DNA melting; anchors Cas9 to the target site. |
| sgRNA Scaffold | Binds the REC and NUC lobes, bridging the complex. | Adopts a pre-ordered T-shaped structure that guides DNA positioning. |
| Target DNA | Provides complementary sequence (protospacer) and PAM. | Undergoes local melting; the displaced non-target strand forms an R-loop. |
Assembly follows an ordered pathway: 1) Cas9 pre-assembles with the sgRNA to form a surveillance complex, 2) The complex scans DNA for a valid PAM via the PI domain, 3) PAM recognition triggers local DNA melting, enabling the sgRNA spacer to interrogate potential complementarity, 4) Full complementarity propagates, inducing full R-loop formation and HNH domain activation, and 5) The catalytically competent complex cleaves both DNA strands.
Diagram 1: Ternary Complex Assembly Pathway
Title: Cas9-sgRNA-DNA Assembly and Activation Pathway
3. Key Experimental Methodologies for Visualization
Understanding this assembly relies on structural and biophysical techniques.
Protocol 3.1: Cryo-Electron Microscopy (Cryo-EM) of the Ternary Complex Objective: Determine high-resolution 3D structure of the assembled Cas9:sgRNA:target DNA complex.
- Sample Preparation: Purify recombinant SpCas9. Synthesize and fold sgRNA. Anneal target DNA oligonucleotide containing a canonical PAM. Incubate components at a 1:1.2:1.5 molar ratio (Cas9:sgRNA:DNA) to form the complex. Apply 3-4 µL of sample (~3 mg/mL) to a plasma-cleaned cryo-EM grid.
- Vitrification: Blot the grid and plunge-freeze in liquid ethane using a vitrification device (e.g., Vitrobot).
- Data Collection: Image grids on a 300 keV cryo-electron microscope equipped with a direct electron detector. Collect 2,000-5,000 micrograph movies at a defocus range of -0.5 to -2.5 µm.
- Image Processing: Perform motion correction and CTF estimation. Pick particles automatically, followed by 2D classification to discard junk. Use an initial model for 3D classification to isolate states (e.g., pre- vs. post-catalytic). Refine the selected class via 3D auto-refinement and Bayesian polishing. Final resolution is typically 3-4 Å.
- Model Building: Fit existing Cas9 crystal structures (PDB: 4ZT0) into the cryo-EM density map using Chimera. Build and refine the sgRNA:DNA heteroduplex and adjust protein sidechains in Coot. Perform real-space refinement in Phenix.
Protocol 3.2: Single-Molecule FRET (smFRET) to Monitor Conformational Dynamics Objective: Observe real-time conformational changes during R-loop formation.
- Labeling: Design a target DNA duplex with Cy3 donor on the 5’ end of the non-target strand and Cy5 acceptor on the 5’ end of the target strand. Purify and anneal DNA.
- Surface Immobilization: Passivate a quartz microfluidic chamber with PEG-biotin. Introduce streptavidin, then biotinylated anti-digoxigenin antibody. Immobilize digoxigenin-labeled Cas9:sgRNA complex.
- Data Acquisition: Introduce labeled DNA in imaging buffer with oxygen scavengers. Illuminate with a 532 nm laser on a TIRF microscope. Acquire donor (Cy3) and acceptor (Cy5) emission movies at 100 ms integration time.
- Analysis: Identify colocalized donor-acceptor spots. Calculate FRET efficiency (E) = IA / (ID + I_A) for each time trace. Plot E over time; low E indicates an open, unpaired state; high E indicates R-loop formation and strand displacement.
Table 2: Key Parameters from Ternary Complex Structural Studies
| Parameter | Cryo-EM Value (SpCas9) | smFRET Observation | Significance |
|---|---|---|---|
| Overall Complex Dimensions | ~100 Å x 110 Å x 50 Å | N/A | Defines molecular footprint for delivery. |
| R-loop Length | ~10 bp (seed) to full 20 bp | Progressive stabilization over 10-200 ms | Kinetics of interrogation dictates specificity. |
| HNH Domain Rotation | ~35° upon activation | Two-state, concerted movement | Correlates directly with catalytic activation. |
| REC Lobe Conformation Change | Significant closure upon binding | Multi-step, induced fit | Essential for discrimination against off-targets. |
Diagram 2: Experimental Workflow for Structural Analysis
Title: Structural & Biophysical Analysis Workflows
4. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Ternary Complex Studies
| Item | Function in Research | Example/Note |
|---|---|---|
| Recombinant Cas9 Nuclease (Wild-type & Variants) | Core protein for in vitro complex formation and structural studies. | Catalytically dead dCas9 is essential for stable complex capture. |
| Chemically Modified sgRNA | Enhances stability and assembly for crystallography/cryo-EM. | 2'-O-methyl, phosphorothioate backbones at 3' terminus. |
| Synthetic DNA Oligonucleotides (with PAM) | For forming target DNA duplexes; site-specific labeling. | HPLC-purified, with modifications (biotin, digoxigenin, fluorophores). |
| Fluorescent Nucleotides (Cy3, Cy5, ATTO dyes) | For smFRET and single-molecule tracking experiments. | Paired with appropriate quenching systems for clean signal. |
| Cryo-EM Grids (Quantifoil, UltrAuFoil) | Supports for vitrified sample in electron microscopy. | Choice of grid type (holey carbon, gold) affects ice quality. |
| Streptavidin & Biotinylated PEG | For surface passivation and complex immobilization in smFRET. | Creates a non-stick surface to prevent non-specific binding. |
| Anti-Digoxigenin Antibody (Biotinylated) | Enforces specific, oriented immobilization of dig-labeled complexes. | Critical for consistent single-molecule data. |
| Oxygen Scavenging System (e.g., PCA/PCD) | Prolongs fluorophore lifespan in single-molecule assays. | Typically protocatechuic acid (PCA) and protocatechuate-3,4-dioxygenase (PCD). |
5. Implications for Drug Development and Protein Engineering
Visualizing the ternary complex at atomic and dynamic levels directly enables rational engineering. Understanding HNH/RuvC positioning supports the development of nickases or FokI-fused dimeric nucleases. Mapping the REC lobe's role in discrimination informs high-fidelity variants (e.g., HypaCas9). The structural blueprint of the assembled complex is crucial for designing anti-CRISPR proteins, guide RNA optimizations, and small-molecule modulators that target specific assembly intermediates for therapeutic control. This structural knowledge, central to domain architecture research, remains the cornerstone of translating CRISPR-Cas9 into precise genetic medicines.
From Structure to Function: Applying Domain Knowledge in Experimental Design and Delivery
sgRNA Design Principles Informed by REC Lobe Interactions and Seed Sequence Positioning
This whitepaper is framed within the context of a broader thesis investigating Cas9 protein domain architecture and structural organization. Understanding the precise spatial arrangement of the Recognition (REC) lobe and Nuclease (NUC) lobe is critical for rational sgRNA design, which directly impacts CRISPR-Cas9 genome editing efficiency and specificity. This guide elucidates how sgRNA architecture, particularly the positioning of the seed sequence (the 10-12 nucleotides proximal to the PAM), is governed by its dynamic interactions with the REC lobe, a key determinant of DNA target strand hybridization and cleavage fidelity.
Structural Basis of REC Lobe-sgRNA Interactions
The REC lobe, primarily comprising the REC1, REC2, and REC3 domains, acts as a molecular scaffold that facilitates the transition of the sgRNA:DNA heteroduplex into an active conformation. Recent structural studies (e.g., cryo-EM and X-ray crystallography) reveal that the REC lobe directly contacts the repeat:antirepeat duplex of the sgRNA scaffold and monitors the correct base-pairing in the seed region.
The following table summarizes critical interaction distances derived from recent high-resolution structural data (PDB IDs: 7OZB, 8F7Z).
Table 1: Key Interatomic Distances in REC Lobe-sgRNA Interface
| Interaction Pair | Average Distance (Å) | Structural Domain Involved | Functional Implication |
|---|---|---|---|
| REC2 (R66) - sgRNA (Phosphate 10) | 2.9 ± 0.3 | REC2 - sgRNA backbone | Stabilizes scaffold architecture |
| REC3 (K510) - sgRNA (Nucleotide -4) | 3.1 ± 0.2 | REC3 - Seed region | Monitors seed hybridization |
| REC1 (H40) - DNA Target Strand (PAM -1) | 4.2 ± 0.5 | REC1 - DNA interface | Positional sensing of PAM distortion |
| Bridge Helix (K848) - sgRNA-DNA Heteroduplex | 3.5 ± 0.4 | BH - Hybrid duplex | Facilitates strand separation |
Seed Sequence Positioning and Energetic Determinants
The seed sequence is positioned within a groove formed by the REC2 and REC3 domains. Optimal positioning is energetically driven, with mismatches in the seed region causing significant distortion and reduced cleavage rates.
Table 2: Impact of Seed Sequence Mismatches on Cleavage Efficiency (Kcat/Km)
| Mismatch Position (from PAM) | Relative Cleavage Efficiency (%) | ΔΔG (kcal/mol) of Binding | Observed REC Lobe Conformational Change |
|---|---|---|---|
| -1 (PAM proximal) | 12 ± 3 | +4.8 ± 0.5 | REC3 domain retraction >8 Å |
| -3 | 28 ± 5 | +3.2 ± 0.4 | Minor REC2 sidechain rearrangement |
| -5 | 65 ± 8 | +1.5 ± 0.3 | No significant structural change |
| -8 | 85 ± 7 | +0.7 ± 0.2 | No significant structural change |
Experimental Protocols for Investigating REC-sgRNA Interactions
Protocol: Cryo-EM Structural Analysis of Cas9-sgRNA-DNA Ternary Complexes
Objective: To resolve the high-resolution structure of the ternary complex to visualize REC lobe interactions.
- Complex Formation: Incubate 5 µM purified S. pyogenes Cas9 with 7.5 µM sgRNA (designed with target of interest) in buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl2) for 10 min at 25°C. Add 10 µM target DNA duplex (containing a 5' NGG PAM) and incubate for further 15 min.
- Grid Preparation: Apply 3.5 µL of complex to a glow-discharged Quantifoil R1.2/1.3 300-mesh Au grid. Blot for 3.5 seconds at 100% humidity and plunge-freeze in liquid ethane using a Vitrobot Mark IV.
- Data Collection: Image grids on a 300 keV cryo-electron microscope (e.g., Titan Krios) equipped with a Gatan K3 direct electron detector. Collect ~5,000 movies at a nominal magnification of 105,000x, corresponding to a pixel size of 0.826 Å, with a total dose of 50 e⁻/Ų.
- Processing: Use RELION or cryoSPARC for motion correction, CTF estimation, particle picking (~2 million), 2D classification, ab initio reconstruction, and high-resolution 3D refinement. Model building into the final map is performed in Coot and refined in Phenix.
Protocol: Single-Molecule FRET to Probe REC Lobe Dynamics
Objective: To measure real-time conformational changes in the REC lobe upon seed mismatch.
- Dye Labeling: Engineer surface-exposed cysteines on the REC2 domain (e.g., S61C) and the NUC lobe (e.g., S867C). Label with maleimide-conjugated FRET pair (Cy3 donor, Cy5 acceptor).
- Immobilization: Biotinylate the 3' end of the target DNA strand and immobilize on a PEG-passivated, streptavidin-coated quartz microfluidic chamber.
- Measurement: Flow in labeled Cas9-sgRNA complexes (with matched or mismatched seed sequences) in imaging buffer (with oxygen scavenger and triplet quencher). Illuminate with a 532 nm laser on a TIRF microscope. Record donor and acceptor emission intensities over time for 100+ individual molecules.
- Analysis: Calculate FRET efficiency (E = IA/(ID+IA)). Plot histograms and identify distinct FRET states corresponding to "open" and "closed" REC lobe conformations.
Diagram: REC Lobe-Guided sgRNA Design Workflow
Title: sgRNA Design Workflow Guided by REC Lobe
Diagram: Cas9-sgRNA-DNA Ternary Complex Architecture
Title: Key Interactions in Cas9-sgRNA-DNA Complex
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for REC Lobe and sgRNA Interaction Studies
| Reagent/Material | Function & Rationale |
|---|---|
| Purified Wild-type & REC Domain Mutant Cas9 | For comparative structural and biochemical assays to dissect domain-specific functions. |
| Chemically Modified sgRNAs (2'-O-Methyl, Phosphorothioates) | To probe backbone interaction points with REC lobe and enhance nuclease stability in functional assays. |
| Fluorophore-labeled Nucleotides (Cy3/Cy5-dUTP) | For incorporation into target DNA for single-molecule FRET experiments monitoring conformational dynamics. |
| Biotinylated DNA Oligos & Streptavidin-coated Beads/Chambers | For immobilization of target DNA in single-molecule or pull-down assays. |
| Crosslinking Agents (Formaldehyde, BS3) | To capture transient REC lobe-sgRNA interactions for structural mass spectrometry. |
| Reconstituted in vitro Transcription/Translation System | For high-throughput screening of sgRNA libraries with Cas9, assessing cleavage kinetics. |
| Next-Generation Sequencing (NGS) Library Prep Kits | For comprehensive profiling of on- and off-target cleavage events (e.g., GUIDE-seq, CIRCLE-seq). |
Informed sgRNA design requires a mechanistic understanding of Cas9's internal architecture, specifically the critical role of the REC lobe in stabilizing the sgRNA scaffold and verifying seed sequence complementarity. By integrating structural data on REC lobe interactions with energetic profiles of seed mismatches, researchers can move beyond empirical rules to rationally engineer sgRNAs with maximal on-target activity and minimal off-target effects. This approach, rooted in structural organization research, is essential for advancing therapeutic genome editing applications, where precision is paramount.
This whitepaper is framed within a broader thesis on Cas9 protein domain architecture and structural organization. The precise recognition of a short Protospacer Adjacent Motif (PAM) by the Cas9 endonuclease is a fundamental determinant of targeting specificity and genome editing efficiency across orthologs. This guide details the structural basis of PAM recognition, quantitative comparisons of ortholog specificity, and experimental protocols for its characterization.
Structural Basis of PAM Recognition
Cas9 orthologs possess distinct PAM Interaction (PI) domains, typically within the C-terminal region, which govern PAM specificity through direct DNA interrogation. The structural constraints of this domain—including its size, charge distribution, and conformational plasticity—dictate the nucleotide sequence recognized.
Table 1: Key Cas9 Orthologs, PAM Specificities, and Structural Features
| Cas9 Ortholog (Source) | Canonical PAM Sequence | PI Domain Key Structural Motifs | Temp. Optima (°C) | Reference (Example) |
|---|---|---|---|---|
| Streptococcus pyogenes (SpCas9) | 5'-NGG-3' | A phosphate lock loop, arginine-rich channel | 37 | Anders et al., 2014 |
| Staphylococcus aureus (SaCas9) | 5'-NNGRRT-3' | Compact β-strand bundle, narrowed groove | 37 | Nishimasu et al., 2015 |
| Campylobacter jejuni (CjCas9) | 5'-NNNNRYAC-3' | Extended α-helical wing, dual recognition loops | 37 | Yamada et al., 2017 |
| Geobacillus stearothermophilus (GeoCas9) | 5'-NNNNCRAA-3' | Stabilized β-sheet core, hydrophobic cleft | 55 | Harrington et al., 2017 |
| Neisseria meningitidis (NmCas9) | 5'-NNNNGATT-3' | Triple-helix bundle, solvent-exposed basic patch | 37 | Lee et al., 2016 |
Experimental Protocols for PAM Characterization
Protocol 1: PAM Depletion Assay (PAMDA)
Purpose: To comprehensively determine the PAM preference of a Cas9 ortholog in vitro. Materials:
- Purified Cas9 protein and sgRNA.
- A randomized PAM library plasmid (e.g., a 10-nt randomized region flanking the protospacer).
- In vitro cleavage reagents (NEBuffer r3.1, MgCl₂). Method:
- Incubate the randomized plasmid library (1 µg) with Cas9:sgRNA RNP complex (100 nM) for 1 hour at 37°C.
- Run the reaction products on an agarose gel. Excise and purify the linearized (cleaved) DNA fraction.
- Amplify the PAM region from the cleaved pool via PCR and submit for high-throughput sequencing.
- Compare the frequency of each PAM sequence in the cleaved pool versus the initial library using bioinformatic tools (e.g., PAMDA analysis pipeline). Enrichment scores indicate preference.
Protocol 2: Structural Validation via X-ray Crystallography
Purpose: To visualize atomic-level interactions between the Cas9 PI domain and its cognate PAM DNA. Method:
- Express and purify the recombinant Cas9 PI domain (or full protein) with an affinity tag.
- Anneal complementary oligonucleotides containing the putative PAM sequence.
- Form the protein-DNA complex by mixing at a 1:1.2 molar ratio. Purify via size-exclusion chromatography.
- Crystallize the complex using vapor diffusion. Screen commercial sparse matrix kits.
- Collect diffraction data at a synchrotron source, solve the structure via molecular replacement, and refine. Analyze PAM-base contacts (hydrogen bonds, van der Waals forces).
Visualizing PAM Recognition and Ortholog Selection
Title: Cas9 Ortholog Selection and PAM Recognition Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PAM Specificity Research
| Reagent / Material | Function / Application | Example Supplier |
|---|---|---|
| PAM Discovery Plasmid Library (e.g., pPAM-Lib) | In vitro randomized library for unbiased PAM profiling. | Addgene (#100000) |
| Recombinant His-tagged Cas9 Orthologs | Purified, active protein for biochemical assays and structural studies. | GenScript (Custom) |
| sgRNA In Vitro Transcription Kit | High-yield synthesis of sgRNA for RNP complex formation. | NEB (E2040S) |
| High-Fidelity DNA Polymerase | Accurate amplification of PAM regions for sequencing libraries. | Thermo Fisher (F-530S) |
| Structure Screen Cryo Kits | Crystallization screening for protein-DNA complexes. | Molecular Dimensions (MD1-46) |
| Next-Gen Sequencing Kit (MiSeq) | Deep sequencing of PAM depletion assay outputs. | Illumina (MS-102-2001) |
| Anti-CRISPR Proteins (e.g., AcrIIA4) | Negative controls to inhibit Cas9 activity and confirm specificity. | ABCAM (ab272255) |
Selecting the optimal Cas9 ortholog for a given genome editing application requires matching the target site's adjacent sequence to the structural constraints of the ortholog's PI domain. Systematic PAM characterization and an understanding of domain architecture are critical for expanding the targeting scope and precision of CRISPR-Cas9 technologies in therapeutic development.
Within the broader research thesis on Cas9 protein domain architecture and structural organization, a critical applied challenge emerges: delivery. The functional unit for genome editing—Cas9 protein plus its guide RNA (sgRNA)—constitutes a large (~160 kDa, ~4.2 kb coding sequence) ribonucleoprotein (RNP) complex. This review provides an in-depth technical guide on exploiting detailed structural knowledge of Cas9 to engineer efficient delivery strategies, categorizing approaches by their reliance on Cas9's size, charge, and domain organization.
Table 1: Key Physical and Functional Parameters of Common Cas9 Orthologs Relevant to Delivery
| Cas9 Ortholog | Protein Size (kDa) | sgRNA Length (nt) | Total RNP Size (MDa, approx.) | Nuclear Localization Signals (NLSs) | Isoelectric Point (pI) |
|---|---|---|---|---|---|
| S. pyogenes (SpCas9) | 158 | ~100 | 3.8-4.2 | Typically 2-4 (C-term &/or N-term) | ~9.0-9.5 (basic) |
| S. aureus (SaCas9) | 105 | ~100 | ~2.7 | 2-3 NLSs common | ~9.3 (basic) |
| C. jejuni (CjCas9) | 112 | ~90 | ~2.6 | 1-2 NLSs | ~8.2 (basic) |
| Campylobacter GeCas9 | 108 | ~110 | ~2.7 | 1-2 NLSs | ~8.5 (basic) |
Leveraging Structural Features for Viral Vector Design
Core Concept: Viral packaging constraints necessitate the use of smaller Cas9 orthologs or split-inteln systems informed by domain boundaries.
3.1. AAV Vector Optimization Based on Size AAV has a ~4.7 kb packaging limit. SpCas9 cDNA (~4.2 kb) leaves minimal space for promoters, sgRNA, and regulatory elements. Strategies include:
- Ortholog Switching: Using smaller Cas9s (e.g., SaCas9, CjCas9).
- Dual-Vector Systems: Splitting Cas9 and sgRNA expression cassettes across two AAVs.
- Intein-Mediated Protein Trans-Splicing: Exploiting Cas9's multi-domain structure to split it at specific loops (e.g., between REC2 and RuvC domains) for reconstitution post-delivery.
Experimental Protocol: Intein-Split AAV Production & Testing
- Split Site Selection: Analyze SpCas9 crystal structure (PDB: 4UN3). Identify solvent-exposed, flexible loops connecting structurally autonomous domains.
- Plasmid Construction: Clone N-terminal fragment (amino acids 1-573) fused to Npu DnaE intein N-half, and C-terminal fragment (574-1368) fused to intein C-half into separate AAV expression plasmids (e.g., pAAV-CB-hybrid promoters).
- Vector Production: Co-transfect HEK293T cells with AAV rep/cap plasmid (serotype, e.g., AAV9), adenoviral helper plasmid, and each AAV transgene plasmid. Purify via iodixanol gradient ultracentrifugion.
- In Vivo Validation: Co-inject both AAVs into mouse tail vein (1e11 vg each). Harvest tissue at 2-4 weeks. Assess:
- Splicing Efficiency: Western blot for full-length Cas9.
- Editing: T7E1/SURVEYOR assay or NGS on genomic DNA.
Diagram Title: Workflow for Intein-Split Cas9 AAV Delivery
Non-Viral Delivery Informed by Surface Charge & Domain Organization
Core Concept: Cas9's highly positive charge (pI ~9.3) facilitates complexation with anionic lipids/polymers but can cause non-specific binding and toxicity. Structural knowledge guides surface engineering.
4.1. Lipid Nanoparticle (LNP) Formulation for Cas9 RNP
- Strategy: Directly encapsulate pre-assembled Cas9 RNP, protecting it from degradation and leveraging endosomal escape capabilities of ionizable lipids.
- Key Consideration: Use of sgRNA with chemical modifications (e.g., 2'-O-methyl, phosphorothioate) to enhance stability, informed by RNP structure showing tolerated modification sites.
Experimental Protocol: LNP Formulation of Cas9 RNP via Microfluidic Mixing
- RNP Preparation: Purify recombinant Cas9. Chemically synthesize and modify sgRNA. Mix at molar ratio of 1:1.2 (Cas9:sgRNA) for 10 min at 25°C.
- LNP Preparation:
- Organic Phase: Ionizable lipid (e.g., DLin-MC3-DMA), DSPC, cholesterol, DMG-PEG in ethanol.
- Aqueous Phase: Cas9 RNP in citrate buffer (pH 4.0).
- Mixing: Use a microfluidic mixer (e.g., NanoAssemblr) with total flow rate (TFR) 12 mL/min and flow rate ratio (FRR, aqueous:organic) 3:1.
- Characterization: Measure particle size (DLS), PDI, and encapsulation efficiency (RiboGreen assay).
- Delivery: Test in vitro on HeLa cells. Analyze editing via NGS 72h post-transfection.
4.2. Cas9 Surface Engineering for Improved Biocompatibility
- PEGylation: Attach PEG to surface-exposed lysines (abundant on Cas9) to reduce aggregation and immunogenicity.
- Targeted Peptide Fusion: Fuse cell-penetrating or targeting peptides (e.g., RGD, transferrin-binding) to Cas9's N- or C-termini, locations distal to the catalytic and sgRNA-binding clefts.
Diagram Title: Engineered Cas9 RNP in Targeted LNP Structure
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Cas9 Delivery Research
| Reagent / Material | Supplier Examples | Function in Delivery Research |
|---|---|---|
| Recombinant S. pyogenes Cas9 Nuclease | Thermo Fisher, Sigma-Aldrich, Horizon Discovery | Gold-standard protein for RNP assembly, in vitro and ex vivo delivery studies. |
| AAV rep/cap & Helper Plasmids (Serotype 2, 6, 9, etc.) | Addgene, Vigene Biosciences | Essential for producing recombinant AAV vectors with specific tropisms. |
| Ionizable Cationic Lipid (e.g., DLin-MC3-DMA, SM-102) | MedChemExpress, Avanti Polar Lipids | Critical component of LNPs for nucleic acid/RNP encapsulation and endosomal escape. |
| Microfluidic Mixer (NanoAssemblr, iLiNP) | Precision NanoSystems, Tecan | Enables reproducible, scalable formulation of LNPs with narrow size distribution. |
| Chemically Modified sgRNA (2'-O-methyl, Phosphorothioate) | Trilink Biotechnologies, Synthego, IDT | Enhances nuclease stability and reduces immunogenicity of RNP complexes. |
| Cell-Penetrating Peptides (e.g., TAT, PF14) | Genscript, AnaSpec | Conjugated to Cas9 or delivery carrier to enhance cellular uptake via non-endocytic pathways. |
| Endosomal Escape Indicator (e.g., LysoTracker, Gal8-mCherry) | Thermo Fisher, Addgene (plasmid) | Fluorescent probes to evaluate the efficiency of endosomal disruption by delivery vectors. |
| Next-Generation Sequencing Kit (for Indel Analysis) | Illumina, Paragon Genomics | For quantitative, unbiased measurement of on-target and off-target genome editing outcomes. |
Effective delivery of CRISPR-Cas9 is not merely a packaging problem but a structural engineering challenge. The size dictates viral cargo limits, the surface charge guides non-viral complexation, and the modular domain architecture enables sophisticated solutions like split proteins. Advancements in delivery will continue to be driven by deep integration of Cas9 structural biology with biomaterials science and vector engineering.
The engineering of CRISPR-Cas9 fusion proteins represents a pivotal advancement in precision genome manipulation, extending beyond simple cleavage to include targeted nucleotide editing and transcriptional regulation. This whitepaper, framed within a broader thesis on Cas9 protein domain architecture and structural organization, examines the critical structural insights required for successfully fusing effector domains—such as cytidine/adenine deaminases for base editing or transcriptional activators/repressors—to the Cas9 scaffold. The core challenge lies in integrating these domains without compromising Cas9's DNA-binding fidelity, effector activity, or cellular delivery efficiency.
Structural Considerations for Effector Domain Fusion
Cas9 Structural Organization and Fusion Sites
The canonical Streptococcus pyogenes Cas9 (spCas9) provides defined termini and internal loops suitable for fusion. Successful fusion depends on maintaining the conformational flexibility required for effector function.
Table 1: Primary Fusion Sites in spCas9 for Effector Domains
| Fusion Site | Structural Location (PDB ID) | Suited Effector Types | Key Structural Constraint |
|---|---|---|---|
| N-terminus | N/A, precedes REC lobe | Large domains (e.g., VP64, p65) | May interfere with REC lobe dynamics for DNA recognition. |
| C-terminus | Follows PAM-interacting domain | Base editor deaminases, compact effectors | Less interference with DNA binding; linker length is critical. |
| Internal Linker (e.g., after residue 713) | Between RuvC and HNH nuclease domains | Deaminases (for base editors) | Requires inactivation of native nuclease activity (D10A, H840A). |
| dCas9 (catalytically dead) Backbone | Entire surface available | Both single and multi-domain effectors | Provides a stable, DNA-targeting scaffold with no cleavage. |
Linker Design Principles
Linkers bridge the Cas9 scaffold and the effector domain. Their design dictates fusion protein performance.
Table 2: Quantitative Analysis of Linker Properties
| Linker Type | Typical Length (AA) | Flexibility (GRAVY Index*) | Common Sequence Motif | Application Example |
|---|---|---|---|---|
| Flexible (Gly-Ser) | 10-30 | Highly Negative (-0.5 to -1.5) | (GGGS)n or (GGGGS)n | Base editor fusions (BE4). |
| Rigid (α-helical) | 12-24 | Variable, often positive | (EAAAK)n | Fusions requiring fixed spacing. |
| Cleavable (e.g., T2A) | 18-22 | N/A | GSGATNFSLLKQAGDVEENPGP | For co-translational separation. |
| *Grand Average of Hydropathicity (GRAVY): More negative values indicate higher hydrophilicity/flexibility. |
Detailed Experimental Protocols
Protocol for Evaluating Fusion Protein Activity via a GFP Reporter Assay
This protocol assesses the functionality of a dCas9-Effector fusion designed for transcriptional activation.
Materials:
- HEK293T cells (ATCC CRL-3216)
- Plasmid constructs: pXPR_023 (dCas9-effector fusion), pU6-gRNA (targeting GFP locus), pGFP-Reporter (with minimal promoter).
- Transfection reagent (e.g., PEI MAX).
- Flow cytometer.
Procedure:
- Day 1: Seed HEK293T cells in a 24-well plate at 1.5 x 10^5 cells/well in DMEM + 10% FBS.
- Day 2: Transfect cells with a total of 1 µg DNA per well using a 3:1:1 mass ratio (pGFP-Reporter : pXPR_023 : pU6-gRNA). Include controls (effector only, gRNA only).
- Day 4 (48h post-transfection): Harvest cells, wash with PBS, and resuspend in FACS buffer.
- Analyze GFP fluorescence intensity for ≥10,000 single-cell events using flow cytometry (e.g., 488 nm excitation, 530/30 nm filter).
- Data Analysis: Calculate mean fluorescence intensity (MFI) for each condition. Fold activation = (MFI sample) / (MFI non-targeting gRNA control).
Protocol for Structural Analysis via SEC-SAXS
Size Exclusion Chromatography coupled with Small-Angle X-ray Scattering (SEC-SAXS) provides solution-state structural insights into fusion protein conformation.
Materials:
- Purified fusion protein (>2 mg/mL in low-salt buffer, e.g., 20 mM HEPES pH 7.5, 150 mM KCl).
- SEC column (e.g., Superose 6 Increase 10/300 GL) pre-equilibrated with matched buffer.
- Synchrotron SAXS beamline or lab-based instrument (e.g., BioXTreme).
Procedure:
- Sample Preparation: Centrifuge protein sample at 16,000 x g for 10 min at 4°C to remove aggregates.
- SEC-SAXS Run: Inject 50 µL of sample onto the SEC column, flowing at 0.5 mL/min directly into the SAXS flow cell.
- Data Collection: Collect 1-second X-ray exposures continuously throughout the elution. Buffer scattering is collected before the void volume.
- Primary Analysis: Use ATSAS software suite. Subtract buffer scattering from the peak frames. Generate the pair-distance distribution function [P(r)] to estimate the maximum particle dimension (Dmax) and radius of gyration (Rg).
- Modeling: Ab initio bead models can be generated using DAMMIF. If a high-resolution structure exists, perform rigid-body modeling of the Cas9 and effector domains.
Diagrams for Signaling Pathways and Workflows
Diagram Title: Base Editor Protein Engineering & Validation Workflow
Diagram Title: dCas9-Effector Transcriptional Activation Mechanism
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Fusion Protein Engineering Research
| Reagent/Material | Supplier Example (Catalogue #) | Function in Research |
|---|---|---|
| pSpCas9(1.1) Plasmid | Addgene (#140032) | Backbone for constructing N- or C-terminal fusions to spCas9. |
| dCas9-VPR Plasmid | Addgene (#114189) | Positive control for transcriptional activation assays. |
| APOBEC1 (rat) cDNA | Addgene (#79620) | Effector domain for creating cytidine base editors. |
| HRV 3C Protease | MilliporeSigma (71493) | For cleaving affinity tags during protein purification. |
| Superose 6 Increase 10/300 GL | Cytiva (29091596) | SEC column for separating folded fusion proteins from aggregates. |
| PEI MAX (40k) | Polysciences (24765) | High-efficiency transfection reagent for delivering large plasmids. |
| KAPA HiFi HotStart ReadyMix | Roche (07958846001) | High-fidelity PCR for amplifying effector domains and linkers. |
| Gibson Assembly Master Mix | NEB (E2611L) | Seamless cloning of multiple fragments (Cas9, linker, effector). |
This analysis is framed within a broader thesis investigating Cas9 protein domain architecture and structural organization. The central premise posits that the functional application of Cas9—whether in a controlled in vitro setting or within the complex milieu of a living cell (in vivo)—imposes distinct and critical structural requirements. These requirements dictate strategic modifications to the core protein architecture to optimize stability, achieve correct subcellular localization, and facilitate the formation of productive ribonucleoprotein (RNP) complexes.
Core Structural Considerations: A Comparative Analysis
The following table summarizes the primary structural and environmental factors differentiating in vitro and in vivo applications.
Table 1: Key Differentiators Between In Vitro and In Vivo Environments
| Consideration | In Vitro Application | In Vivo Application |
|---|---|---|
| Primary Stability Concern | Thermostability, shelf-life, freeze-thaw cycles. | Proteolytic degradation, thermal denaturation at 37°C, oxidative stress. |
| Localization Requirement | Not applicable (homogenous solution). | Nuclear import (for DNA targeting), organelle-specific targeting (mitochondria, chloroplast). |
| Complex Formation | Direct assembly of purified Cas9 and sgRNA. | Delivery and intracellular assembly of Cas9 and sgRNA components; competition with cellular RNA/DNA-binding proteins. |
| Cellular Environment | Defined buffer (controlled pH, salts, Mg²⁺). | Crowded, reducing environment, variable pH, nucleases, proteases, immune sensors. |
| Key Structural Modifications | Point mutations for thermostability (e.g., Geobacillus sp. Cas9). | Fusion with Nuclear Localization Signals (NLSs), degradation-resistant motifs, deimmunizing mutations. |
Detailed Methodologies for Key Experiments
Protocol 1: Assessing Thermostability via Differential Scanning Fluorimetry (DSF)
- Objective: To quantify the melting temperature (Tm) of wild-type and engineered Cas9 variants for in vitro use.
- Procedure:
- Purify Cas9 protein and dialyze into a storage buffer (e.g., 20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT).
- Prepare a 5X concentrated solution of a fluorescent dye (e.g., SYPRO Orange) in the same buffer.
- In a 96-well PCR plate, mix 20 µL of 2 µM Cas9 protein with 5 µL of 5X dye. Include buffer-only controls.
- Seal the plate and centrifuge briefly.
- Run the DSF assay on a real-time PCR instrument using a temperature ramp from 25°C to 95°C at a rate of 1°C/min, with fluorescence measurements taken continuously.
- Analyze the data by plotting the negative first derivative of fluorescence versus temperature. The peak corresponds to the protein's Tm.
Protocol 2: Evaluating Nuclear Localization Efficiency via Fluorescence Microscopy
- Objective: To validate the function of NLS fusions on Cas9 for in vivo applications.
- Procedure:
- Construct plasmids encoding Cas9 fused to C-terminal, N-terminal, or bipartite NLS sequences (e.g., SV40 NLS, c-Myc NLS) and a fluorescent tag (e.g., EGFP).
- Transfect HEK293T cells cultured on glass-bottom dishes with the constructed plasmids using a standard transfection reagent.
- 24-48 hours post-transfection, stain cell nuclei with Hoechst 33342 (1 µg/mL) for 15 minutes at 37°C.
- Wash cells twice with PBS and image using a confocal fluorescence microscope.
- Quantify localization by measuring the fluorescence intensity ratio of the nucleus to the cytoplasm for at least 50 cells per construct.
Protocol 3: Analyzing RNP Complex Formation via Electrophoretic Mobility Shift Assay (EMSA)
- Objective: To compare the in vitro assembly kinetics and stability of Cas9:sgRNA RNPs.
- Procedure:
- In vitro transcription: Generate a target DNA fragment (200-500 bp) containing the protospacer adjacent motif (PAM) and target sequence.
- RNP Assembly: Pre-complex purified Cas9 with a chemically modified or unmodified sgRNA at a 1:1.2 molar ratio in assembly buffer (20 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl₂, 1 mM DTT) for 10 min at 25°C.
- Binding Reaction: Incubate the pre-assembled RNP with the target DNA fragment (labeled with Cy5) for 30 min at 37°C.
- Electrophoresis: Load reactions onto a 6% native polyacrylamide gel in 0.5X TBE buffer. Run at 100V for 60-90 min at 4°C.
- Detection: Visualize the gel using a fluorescence imager. A shift in the DNA band to a higher molecular weight indicates successful RNP formation and DNA binding.
Essential Diagrams
Diagram 1: Structural Modification Pathways for Application Goals (100 chars)
Diagram 2: Intracellular Pathways for Cas9 Activation (93 chars)
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Structural-Functional Analysis of Cas9
| Item | Function & Relevance |
|---|---|
| High-Purity, Nuclease-Free Cas9 Protein | Essential baseline reagent for in vitro assays (EMSA, DSF) and for forming pre-assembled RNPs for delivery. Purity is critical to avoid off-target effects. |
| Chemically Modified sgRNA (2'-O-methyl, phosphorothioate) | Enhances nuclease resistance in vivo, improving RNP stability and half-life. Critical for in vivo efficacy, less critical for standard in vitro use. |
| Nuclear Localization Signal (NLS) Peptides/Conjugates | Used to validate or enhance nuclear import. Can be fused genetically or chemically conjugated to Cas9 protein for in vivo applications. |
| Protease Inhibitor Cocktails | Used in in vivo-mimicking lysate assays or during protein purification from cells to assess and prevent Cas9 degradation, informing stability engineering. |
| Fluorescent Protein/Epitope Tag Plasmids (e.g., EGFP, HA, FLAG) | Enable tracking of Cas9 localization (microscopy), purification (immunoprecipitation), and quantification (flow cytometry) in cellular environments. |
| SYPRO Orange Dye | A environmentally sensitive fluorescent dye used in DSF assays to measure protein thermal unfolding and determine melting temperature (Tm). |
| Native Gel Electrophoresis System | For EMSAs to visualize Cas9:sgRNA:DNA ternary complex formation and assess binding affinity under different buffer/ modification conditions. |
Addressing Structural Limitations: Strategies for Enhancing Specificity and Efficiency
Within the broader thesis investigating Cas9 protein domain architecture and structural organization, this whitepaper delves into a critical determinant of CRISPR-Cas9 fidelity: the structural basis for DNA mismatch tolerance. High-fidelity Cas9 variants often feature mutations in the REC (recognition) and NUC (nuclease) lobes, underscoring their role in discriminatory proofreading. Off-target effects, a major hurdle in therapeutic and research applications, are directly linked to how these lobes accommodate mismatches between the guide RNA (gRNA) and target DNA. This document synthesizes current structural and biochemical data to elucidate the mechanistic roots of mismatch tolerance.
Structural Organization of Cas9 Lobes
The Streptococcus pyogenes Cas9 (SpCas9) is a bilobed architecture. The REC lobe (comprising REC1, REC2, and REC3 domains) is primarily responsible for gRNA binding and DNA interrogation. The NUC lobe harbors the HNH and RuvC nuclease domains, along with the PI (PAM-interacting) domain. DNA binding induces a conformational change from an inactive to an active state. Mismatches, depending on their position and identity, are sensed through a network of interactions within and between these lobes, affecting the stability of the DNA-RNA heteroduplex and the activation trajectory of the nuclease domains.
Quantitative Analysis of Mismatch Tolerance
Recent high-throughput sequencing studies and single-molecule FRET experiments quantify the impact of mismatches on cleavage efficiency and kinetics. Tolerance is highly position-dependent, with mismatches distal to the PAM (PAM-distal) often better tolerated than those near the PAM (PAM-proximal), particularly in the "seed" region (positions 1-10 from PAM). The data below summarizes key findings from systematic mismatch profiling.
Table 1: Cleavage Efficiency Tolerance to Single Mismatches by Position (Relative to Wild-Type)
| Mismatch Position (PAM-proximal = 1) | Average Cleavage Efficiency (%) | Notes |
|---|---|---|
| 1-5 (Seed Region) | 5-20% | Severe reduction; high-fidelity checkpoint. |
| 6-10 | 10-40% | Moderate tolerance, varies by base identity. |
| 11-15 | 30-70% | Higher tolerance; REC2/REC3 interactions key. |
| 16-20 (PAM-distal) | 50-95% | Often well-tolerated; major role for REC1. |
Table 2: Impact of Lobe-Specific High-Fidelity Mutations on Mismatch Tolerance
| Cas9 Variant | Key Mutations (Lobe) | Reduction in Off-Target Cleavage (Fold) | Notes on Structural Mechanism |
|---|---|---|---|
| SpCas9-HF1 | N497A, R661A, Q695A, Q926A (REC/NUC) | ~10-100x | Reduces non-specific DNA contacts, stabilizes inactive state. |
| eSpCas9(1.1) | K848A, K1003A, R1060A (NUC) | ~10-100x | Alters electrostatic balance, destabilizes mismatched duplex. |
| HypaCas9 | N692A, M694A, Q695A, H698A (REC3) | ~100x | Tightens REC3 "lid", prevents activation with mismatches. |
Experimental Protocols for Assessing Mismatch Tolerance
High-Throughput Mismatch Profiling (GUIDE-seq/Digenome-seq)
Objective: Genome-wide identification of off-target sites with mismatches. Protocol:
- Transfection: Deliver SpCas9-gRNA ribonucleoprotein (RNP) complex into cultured human cells (e.g., HEK293T) alongside a double-stranded oligonucleotide tag (GUIDE-seq tag).
- Tag Integration: Upon double-strand break (DSB), the tag is integrated into genomic sites via non-homologous end joining (NHEJ).
- Genomic DNA Extraction & Processing: Harvest cells after 72 hours. Extract genomic DNA and shear by sonication.
- Library Preparation & Sequencing: Add sequencing adapters, perform PCR enrichment targeting integrated tags, and sequence on an Illumina platform.
- Bioinformatic Analysis: Map sequence reads to the reference genome to identify all tag integration sites, which correspond to DSB locations. Compare to in silico predicted off-target sites with mismatches.
Single-Molecule FRET (smFRET) to Probe Conformational Dynamics
Objective: Measure real-time conformational changes in Cas9 upon binding matched vs. mismatched DNA. Protocol:
- Labeling: Engineer cysteines into specific REC and NUC lobe domains (e.g., REC3 and HNH). Label with donor (Cy3) and acceptor (Cy5) fluorophores.
- Surface Immobilization: Immobilize dye-labeled Cas9:gRNA complexes on a passivated quartz microscope slide via a biotin-streptavidin linkage.
- Data Acquisition: Use a total internal reflection fluorescence (TIRF) microscope. Introduce flowing solutions containing matched or mismatched target DNA duplexes.
- FRET Trace Analysis: Monitor FRET efficiency (proportional to distance between dyes) over time for hundreds of individual molecules. Compare the rates and probabilities of transitioning from low-FRET (inactive) to high-FRET (active, DNA-cleavage competent) states between matched and mismatched substrates.
Visualizing the Mismatch Sensing Pathway
Diagram 1: Cas9 Mismatch Sensing & Activation Decision
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Studying Cas9 Mismatch Tolerance
| Reagent / Material | Function & Application in Research |
|---|---|
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1, HypaCas9) | Engineered proteins with reduced off-target activity; used as comparative controls to wild-type SpCas9 to isolate structural determinants of fidelity. |
| Chemically Modified gRNAs (2'-O-Methyl, Phosphorothioate) | Enhance nuclease stability and can influence mismatch discrimination; useful for probing the role of gRNA backbone interactions with the REC lobe. |
| Fluorophore-Labeled dNTPs (Cy3-dUTP, Cy5-dCTP) | Essential for generating fluorescently labeled DNA substrates for smFRET or gel-based binding/cleavage assays. |
| Biotinylated DNA Oligos & Streptavidin-Coated Surfaces/Beads | For immobilizing DNA substrates in single-molecule experiments or for pull-down assays to measure binding affinity of Cas9 to mismatched targets. |
| Structure-Guided Cas9 Mutant Libraries (REC3, PI domain) | Plasmid collections for saturation mutagenesis to systematically test the functional impact of specific residues on mismatch tolerance. |
| Cell Lines with Reporter Constructs (eGFP disruption, SURVEYOR assays) | Rapid functional readouts for on-target vs. off-target cleavage efficiency in a cellular context. |
| Next-Generation Sequencing Kits (Illumina Compatible) | For GUIDE-seq, CIRCLE-seq, or other high-throughput off-target profiling methods to generate genome-wide mismatch tolerance data. |
| Anti-Cas9 Monoclonal Antibodies | For immunoprecipitation (ChIP-seq) to map Cas9 binding sites genome-wide, including mismatched, non-cleaved engagements. |
This technical guide is framed within a broader research thesis on Cas9 protein domain architecture and structural organization. The central thesis posits that a comprehensive, structure-guided understanding of the spatial and functional arrangement of Cas9 domains—Rec I, Rec II, Rec III, HNH, RuvC, PI, and WED—enables the rational engineering of high-fidelity variants. By systematically targeting residues involved in non-catalytic, DNA backbone interactions, particularly those mediating off-target binding, we can decouple specificity from on-target activity. This document details the principle and execution of this approach, exemplified by pioneering variants like SpCas9-HF1 and eSpCas9.
Structural Foundations for Rational Design
The wild-type Streptococcus pyogenes Cas9 (SpCas9) engages target DNA via a complex network of interactions. Beyond the catalytic HNH (cleaves the target strand) and RuvC (cleaves the non-target strand) domains, numerous non-catalytic domains form hydrogen bonds with the DNA phosphate backbone. The thesis-driven insight is that these energetically additive, non-specific contacts stabilize both on- and off-target complexes. Mutating these residues selectively destabilizes mismatched off-target complexes while preserving sufficient energy for on-target cleavage.
Key Structural Domains and Targetable Interactions:
- REC Lobe (Rec I-III): Responsible for sgRNA and DNA recognition.
- NUC Lobe (HNH, RuvC, PI, WED): Contains catalytic centers and DNA interaction interfaces.
- Target Interaction Residues: Residues in the REC III (e.g., N497, R661), PI (e.g., Q926), and WED (e.g., K848) domains form hydrogen bonds with the DNA phosphate backbone.
High-Fidelity Variants: Design Principles & Comparative Analysis
Two primary strategies emerged from domain-structure analysis:
- Electrostatic Destabilization (eSpCas9): Reduces positive charge in the non-target strand groove (RuvC-III domain) to decrease non-specific interactions with the negatively charged DNA backbone.
- Hydrogen Bond Elimination (SpCas9-HF1): Systematically alanine-substitutes key residues involved in phosphate-backbone hydrogen bonding outside the catalytic centers.
Table 1: Rational Design and Performance of High-Fidelity SpCas9 Variants
| Variant | Underlying Principle | Key Mutations (Domain) | Proposed Effect | On-Target Efficiency (vs. wtSpCas9)* | Off-Target Reduction (vs. wtSpCas9)* |
|---|---|---|---|---|---|
| eSpCas9(1.1) | Weaken non-catalytic DNA binding (Electrostatic) | K848A (WED), K1003A (RuvC-III), R1060A (RuvC-III) | Reduces non-specific groove binding | ~70-90% | 10- to 100-fold+ |
| SpCas9-HF1 | Eliminate specific backbone H-bonds | N497A (Rec III), R661A (Rec III), Q695A (Rec III), Q926A (PI) | Removes stabilizing phosphate contacts | ~60-80% | Undetectable for many sites |
| HypaCas9 | Enhance conformational proofreading | N692A, M694A, Q695A (Rec III), H698A (Rec III) | Stabilizes inactive HNH conformation | ~50-70% | >100-fold for certain sites |
| evoCas9 | Directed evolution from HF1 scaffold | Includes HF1 mutations + additional (e.g., C80R) | Improves fidelity & retains activity | ~70-100% | >10,000-fold in model systems |
| Sniper-Cas9 | Library screening & structure guide | F539S (Rec II), M763I (Unknown), K890N (RuvC) | Optimizes kinetic discrimination | ~80-120% | 10- to 100-fold+ |
*Representative ranges from primary literature; actual performance is highly sequence-context dependent.
Experimental Protocols for Fidelity Assessment
A robust assessment of HiFi variants requires multiple complementary assays.
Protocol 4.1: In Vitro Biochemical Cleavage Assay (GUIDE-seq & BLISS)
Purpose: To quantitatively compare cleavage kinetics and specificity under controlled conditions. Steps:
- Protein Purification: Express and purify wild-type and variant Cas9 proteins (with His/Strep tags) from E. coli using affinity (Ni-NTA/Strep-Tactin) and size-exclusion chromatography.
- Target DNA Preparation: Generate PCR-amplified or plasmid DNA containing the on-target and known/potential off-target sequences.
- Reaction Setup: Pre-complex Cas9 protein with sgRNA (1:1.2 molar ratio) for 10 min at 25°C. Incubate RNP complex with target DNA in cleavage buffer (20 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl2, 5% glycerol).
- Kinetics & Specificity: Perform time-course reactions (e.g., 0, 1, 5, 15, 60 min) at 37°C. Quench with EDTA and Proteinase K.
- Analysis: Resolve cleavage products via agarose or TBE-Urea PAGE. Quantify using gel densitometry. Calculate cleavage rates (k_obs) and mismatch tolerance profiles.
Protocol 4.2: Cell-Based, Genome-Wide Off-Target Detection (CHIP-seq & Digenome-seq)
Purpose: To identify and quantify off-target sites in a living cellular context. Steps (for Digenome-seq):
- Genomic DNA Isolation: Extract high-molecular-weight genomic DNA from untreated human cells.
- In Vitro Digestion: Incubate genomic DNA (1 µg) with saturating RNP complexes (200-400 nM) in cleavage buffer for 24 hours at 37°C.
- Whole-Genome Sequencing (WGS): Fragment the DNA, prepare sequencing libraries, and perform high-coverage WGS (~50-100x).
- Bioinformatic Analysis: Map sequencing reads to the reference genome. Use algorithms (e.g., Digenome-seq tool, Cas-OFFinder) to identify sites with significant cleavage-induced breaks, comparing signal between treated and control DNA.
- Validation: Confirm top off-target sites via targeted deep sequencing (amplicon-seq).
Diagram Title: HiFi Cas9 Engineering & Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Engineering & Testing HiFi Cas9 Variants
| Item | Function & Rationale |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | For error-free amplification of Cas9 expression plasmids and site-directed mutagenesis. |
| Site-Directed Mutagenesis Kit | Introduces specific point mutations into the cas9 gene for variant creation. |
| E. coli Expression Strain (e.g., Rosetta2/ BL21) | Provides optimal tRNAs and background for high-yield, soluble Cas9 protein expression. |
| Nickel-NTA or Strep-Tactin Affinity Resin | Purifies His- or Strep-tagged Cas9 proteins via affinity chromatography. |
| Size-Exclusion Chromatography Column (e.g., Superdex 200) | Polishes purified Cas9 protein, removing aggregates and ensuring monodispersity. |
| In Vitro Transcription Kit (T7) | Produces high-quality, sgRNA for biochemical assays and RNP complex formation. |
| Synthetic Target DNA Oligos & Plasmid Substrates | Serve as defined cleavage targets for in vitro kinetic and specificity assays. |
| Deep Sequencing Platform (e.g., Illumina) | Enables genome-wide, unbiased identification of off-target sites (GUIDE-seq, CIRCLE-seq). |
| Cas9 Off-Target Prediction Software (e.g., Cas-OFFinder) | Computationally predicts potential off-target sites for guide RNA designs. |
| Cell Line with Reportable Loci (e.g., HEK293T with integrated GFP) | Allows rapid, quantitative assessment of on-target editing efficiency in cells. |
Within the broader thesis of Cas9 protein domain architecture and structural organization, the central mechanistic question for function is how conformational rearrangements, dictated by domain organization, mediate the critical transition from target DNA search to cleavage. This guide dissects the precise balance between two indispensable and sequential processes: R-loop formation (DNA unwinding) and catalytic activation of the HNH and RuvC nuclease domains. Optimizing overall cleavage efficiency hinges on understanding and experimentally manipulating this balance, which is governed by allosteric communication between spatially distinct domains.
Core Mechanism: Unwinding to Activation Cascade
Target DNA recognition by the Cas9-sgRNA complex triggers local DNA unwinding, initiating heteroduplex formation between the sgRNA guide strand and the target DNA (the R-loop). Successful R-loop propagation acts as an allosteric signal, inducing large-scale conformational changes that reposition the HNH domain from a solvent-exposed, inactive state to one that engages the DNA target strand. This repositioning, in turn, facilitates the catalytic maturation of the RuvC domain for cleaving the non-target strand. The efficiency of the entire process is rate-limited by the slowest step, often the HNH domain transition.
Diagram Title: Cas9 Cleavage Activation Cascade
Key Quantitative Parameters & Their Measurement
The efficiency balance can be quantified through specific kinetic and biochemical measurements.
Table 1: Key Quantitative Parameters for Assessing Cleavage Balance
| Parameter | Description | Typical Measurement Method | Impact on Efficiency |
|---|---|---|---|
| R-loop Formation Rate (k_R-loop) | Rate of target strand hybridization & non-target strand displacement. | Single-molecule FRET, stopped-flow. | Slow rate creates a kinetic bottleneck. |
| HNH Activation Rate (k_HNH) | Rate of HNH domain conformational switch to active state. | smFRET, time-resolved crystallography. | Often the rate-limiting step post-unwinding. |
| Cleavage Fidelity (ΔΔG) | Free energy difference between on-target and off-target binding/unwinding. | Biochemical competition assays, NGS-based profiling. | Tighter balance favors on-target specificity. |
| Processivity (P_cleave) | Probability that a successful R-loop leads to DSB. | Single-turnover kinetic assays. | Direct measure of coupling efficiency. |
| Domain Mutagenesis Effects (Δk) | Change in rate constants from domain interface mutations. | Comparative enzyme kinetics. | Identifies allosteric communication hubs. |
Experimental Protocols for Decoupling Unwinding from Catalysis
Protocol 4.1: Single-Molecule FRET to Monitor R-loop Dynamics & HNH Movement
- Objective: Simultaneously measure DNA unwinding (R-loop) and HNH domain conformation in real-time.
- Materials: See "Scientist's Toolkit" below.
- Method:
- Dye Labeling: Construct target DNA with Cy3 donor on displaced non-target strand and Cy5 acceptor on the proximal end of the HNH domain (via engineered cysteine).
- Surface Immobilization: Immobilize biotinylated DNA constructs on a neutravidin-coated flow cell.
- Complex Formation: Introduce Cas9-sgRNA (pre-loaded with nontarget strand-blocking DNA oligonucleotide to prevent cleavage) in imaging buffer.
- Data Acquisition: Observe using a TIRF microscope. Initial high FRET (dyes close) indicates pre-catalytic state. R-loop formation causes a FRET decrease (strand separation). Subsequent HNH movement may cause a second FRET change.
- Analysis: Trace individual molecules to calculate rates
k_R-loopandk_HNHand determine their correlation.
Protocol 4.2: Pre-Cleavage Structural Trapping for Cryo-EM Analysis
- Objective: Obtain high-resolution structures of intermediates to define domain architecture at distinct steps.
- Method:
- Complex Assembly: Incubate Cas9-sgRNA with target DNA containing a non-cleavable phosphorothioate modification at the scissile phosphate.
- Crosslinking: Treat the complex with a mild crosslinker (e.g., glutaraldehyde at low concentration) to trap transient conformations.
- Purification: Purify the trapped complex via size-exclusion chromatography.
- Grid Preparation & Imaging: Prepare cryo-EM grids, collect data, and perform 3D classification to separate structural states (e.g., partially unwound, fully unwound pre-HNH flip, post-HNH flip pre-cleavage).
- Analysis: Model domain positions, focusing on HNH-RuvC interface and DNA groove interactions.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Cleavage Balance Studies
| Reagent / Material | Function in Experiment |
|---|---|
| High-Purity, Site-Specifically Labeled Cas9 (e.g., S. pyogenes) | Enables attachment of fluorescent dyes or other probes for smFRET or crosslinking without perturbing activity. |
| Chemically Modified sgRNA (e.g., 3'-biotin, internal dyes) | For complex immobilization or direct observation of RNA dynamics. |
| Synthetic DNA Substrates with Modifications | Non-cleavable (phosphorothioate) or mismatch-containing targets to trap intermediates; fluorescently labeled for unwinding assays. |
| Allosteric Inhibitor/Effector Molecules (e.g., Acr proteins, small molecules) | Tools to perturb specific steps (unwinding vs. activation) to probe their individual contributions to the rate-limiting step. |
| Stoichiometric Cleavage Assay Buffer (e.g., with Ca²⁺) | Divalent cation substitution (Ca²⁺ for Mg²⁺) allows DNA binding and R-loop formation but inhibits catalysis, trapping pre-cleavage states. |
Optimization Strategies: Manipulating the Balance
Understanding this balance allows for rational engineering.
Table 3: Optimization Approaches Based on Mechanism
| Target Process | Strategy | Expected Outcome |
|---|---|---|
| Accelerating Unwinding | Engineered Cas9 variants with positively charged residues in the REC lobe or altered PAM-interacting domain. | Increased k_R-loop, beneficial for targets with high secondary structure. |
| Stabilizing HNH Activation | Mutations that destabilize the HNH auto-inhibitory conformation or strengthen its interface with the R-loop. | Increased k_HNH, improving processivity (P_cleave) and overall speed. |
| Tightening Allosteric Coupling | Directed evolution for variants where RuvC cleavage is strictly dependent on full HNH activation. | Dramatically improved specificity, as partial R-loops (off-targets) fail to trigger DSB. |
| Decoupling for Nickase Generation | Targeted point mutations (D10A for RuvC, H840A for HNH) to study isolated domain function. | Creates tools for precise single-strand break generation or base editing. |
Diagram Title: Factors Influencing the Cleavage Balance
Optimizing Cas9-mediated cleavage is not a singular focus on maximizing catalytic rate but requires a systems-level understanding of the sequential, allosterically gated steps from DNA unwinding to domain activation. Research within the overarching thesis of domain architecture reveals that strategic perturbations at domain interfaces can rebalance this kinetic pathway, enabling the generation of next-generation editors with tailored properties—from ultra-fast to hyper-precise—for advanced therapeutic and research applications.
This whitepaper addresses a critical bottleneck in CRISPR-Cas9 genome editing: the strict requirement for a Protospacer Adjacent Motif (PAM) sequence adjacent to the target DNA. Within the broader thesis research on Cas9 protein domain architecture and structural organization, this work focuses on the PAM-Interacting (PI) domain. The inherent specificity of the wild-type PI domain, while crucial for bacterial immunity, severely limits the targetable genomic loci for therapeutic and research applications. This document provides an in-depth technical guide on the rational and combinatorial structural engineering of the PI domain to relax PAM specificity, thereby expanding the targeting range of CRISPR-Cas9 systems.
Structural Organization of Cas9 and the PI Domain
Cas9 is a multi-domain enzyme. The structural thesis context posits that its function is modularly organized:
- Recognition Lobe (REC): Comprises REC1, REC2, and REC3 domains, responsible for sgRNA binding and target DNA interrogation.
- Nuclease Lobe (NUC): Contains the HNH and RuvC nuclease domains, which cleave the target and non-target DNA strands, respectively.
- PAM-Interacting Domain (PI): A critical component within the NUC lobe, often comprising a PI helix and loop, that directly contacts and recognizes the PAM sequence on the non-target DNA strand.
The PI domain acts as a molecular gatekeeper; its structure dictates which PAM sequences are recognized, thereby licensing subsequent DNA unwinding and cleavage. Engineering this domain is therefore the most direct route to altering PAM specificity.
Quantitative Analysis of Natural and Engineered PAM Specificities
Table 1: PAM Specificities of Wild-Type and Representative Engineered Cas9 Variants
| Cas9 Variant | Origin / Engineering Method | Canonical PAM (Wild-type) | Engineered/Relaxed PAM | Key Structural Alteration(s) in PI Domain | Reference (Example) |
|---|---|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | 5'-NGG-3' | N/A | N/A | Jinek et al., 2012 |
| SpCas9-VQR | SpCas9, Structure-Guided | NGG | 5'-NGAN-3', 5'-NGNG-3' | D1135V, R1335Q, T1337R (PI loop/helix) | Kleinstiver et al., 2015 |
| SpCas9-SpRY | SpCas9, Phage-Assisted Evolution | NGG | 5'-NRN > 5'-NYN-3' (R=A/G, Y=C/T) | A combination of >20 mutations across PI & REC domains | Walton et al., 2020 |
| ScCas9 | Streptococcus canis | 5'-NNG-3' | (Natural variant) | Natural sequence variation in PI domain compared to SpCas9 | Chatterjee et al., 2018 |
| xCas9(3.7) | SpCas9, Phage-Assisted Evolution | NGG | 5'-NG-3', 5'-GAA-3', 5'-GAT-3' | E1219V, D1332A, etc. (Primarily PI domain) | Hu et al., 2018 |
| SpG | SpCas9, Phage-Assisted Evolution | NGG | 5'-NGN-3' | A combination of mutations in the PI domain | Walton et al., 2020 |
Core Methodologies for Engineering the PI Domain
Structure-Guided Rational Design
Protocol: This approach requires a high-resolution crystal or cryo-EM structure of Cas9 bound to DNA containing the PAM.
- Identify Key Residues: Map the residues in the PI domain (e.g., SpCas9 residues 1100-1400) that make direct hydrogen bonds or van der Waals contacts with the PAM nucleotides.
- Molecular Modeling & Docking: Use software like Rosetta, PyMOL, or MODELLER to model amino acid substitutions at these contact positions. Screen in silico for mutations that could potentially accommodate alternative nucleotide bases (e.g., modeling a Glu->Ala change to remove a steric clash with a Guanine, allowing Cytosine binding).
- Saturation Mutagenesis of Hotspots: For residues deemed critical, perform site-saturation mutagenesis libraries focused on the PI helix/loop.
- Library Screening: Clone the mutant library into a bacterial expression system and pair with a negative selection reporter (e.g., a toxin gene with the desired new PAM) and a positive selection reporter (e.g., an antibiotic resistance gene with the canonical PAM) to identify mutants with altered specificity.
Phage-Assisted Continuous Evolution (PACE)
Protocol: This powerful method directs the evolution of Cas9 variants with relaxed PAM requirements in E. coli.
- Construct Mutagenesis Plasmid (MP): Clone the Cas9 gene (with PI domain) into a plasmid with a mutagenesis cassette (e.g., expressing error-prone polymerase genes).
- Set Up Selection Phage: Engineer an M13 bacteriophage to carry:
- A positive selection gene (e.g., gene III essential for infection) under the control of a promoter activated only by functional Cas9-sgRNA cleavage at a relaxed-PAM site.
- A negative selection gene (e.g., a toxin) under the control of a promoter activated by cleavage at the canonical PAM site.
- Run PACE: Infect E. coli harboring the MP with the selection phage in a continuous flow chemostat. Over 100-200 hours of serial infection, only bacterial cells expressing evolved Cas9 variants that cleave the new PAM (activating gene III) and avoid the old PAM (avoiding toxin) will produce progeny phage.
- Isolate & Characterize: Sequence the Cas9 gene from evolved phage pools and individual clones to identify accumulated mutations, which are frequently concentrated in the PI domain.
Key Signaling and Engineering Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PI Domain Engineering Experiments
| Reagent / Material | Function / Purpose in PI Domain Engineering |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | For accurate amplification of Cas9 gene fragments during cloning and library construction. |
| Site-Directed Mutagenesis Kit | For introducing specific point mutations into the PI domain in rational design approaches. |
| Error-Prone PCR Kit | To generate random mutation libraries within the PI domain coding sequence for directed evolution. |
| Bacterial Two-Hybrid or Split-Protein Reporter Systems | To rapidly screen PI domain mutants for PAM recognition specificity via transcriptional activation of a reporter gene (e.g., GFP, LacZ). |
| Negative Selection Toxin Genes (e.g., ccdB, sacB) | Cloned behind canonical PAM sites to select against Cas9 variants that retain original specificity. |
| Positive Selection Genes (e.g., antibiotic resistance, M13 gene III) | Cloned behind desired relaxed PAM sites to select for Cas9 variants with new specificity. |
| Phage-Assisted Continuous Evolution (PACE) Apparatus | Specialized chemostat system for continuous bacterial culture and phage propagation required for PACE experiments. |
| Next-Generation Sequencing (NGS) Platform | For deep sequencing of mutant libraries to identify enriched mutations and characterize PAM preferences (e.g., PAM-SCAN). |
| Recombinant Cas9 Protein (Wild-type & Mutant) | For in vitro biochemical assays (e.g., gel shift EMSA, cleavage assays) to quantitatively measure PAM binding affinity and cleavage kinetics. |
| Cryo-Electron Microscopy (Cryo-EM) Supplies | Grids, vitrification devices, and access to a high-end microscope to solve structures of engineered Cas9 mutants complexed with novel PAM DNA. |
Validation and Characterization of Engineered Variants
After obtaining PI domain mutants, comprehensive validation is essential:
- PAM-SCAN Assay: Clone a randomized PAM library (e.g., NNNN) into a reporter plasmid. Transfert cells with the engineered Cas9 and a targeted sgRNA, then sequence the surviving (uncut) plasmid pool to determine which PAM sequences are no longer recognized/cleaved.
- In Vitro Cleavage Assays: Purify the engineered Cas9 protein and test cleavage efficiency on synthetic DNA substrates containing a spectrum of PAM sequences. Quantify kinetics and efficiency.
- Cellular Activity Profiling: Test the variant's gene editing efficiency (indel formation) at multiple endogenous genomic loci bearing the new PAM sequence, comparing it to wild-type activity at canonical PAM loci.
- Structural Validation: Solve the structure of the engineered Cas9 bound to DNA containing the relaxed PAM. This confirms the predicted structural changes and provides a basis for further rounds of engineering.
The structural engineering of the Cas9 PI domain represents a premier example of how deep understanding of protein domain architecture informs transformative biotechnology. This research, as a core chapter of the broader thesis, demonstrates that the PI domain is a malleable module whose specificity can be reprogrammed through rational and evolutionary strategies. Successfully relaxed PAM specificity, as achieved by variants like SpRY and SpG, removes a fundamental limitation of CRISPR-Cas9, paving the way for more versatile genome editing, synthetic biology, and therapeutic development. Future work will focus on further refining these engineered domains for ultimate specificity, minimal off-target effects, and efficient delivery in vivo.
The therapeutic application of CRISPR-Cas9 is fundamentally constrained by the size of the canonical Streptococcus pyogenes Cas9 (SpCas9, ~1368 amino acids). This large size impedes efficient packaging into viral delivery vectors, such as adeno-associated viruses (AAVs), which have a cargo capacity of ~4.7 kb. This review, framed within a broader thesis on Cas9 protein domain architecture and structural organization, posits that understanding and exploiting the modularity of Cas9 is key to overcoming delivery barriers. Two primary strategies have emerged: 1) mining natural orthologs with compact architectures, and 2) engineering artificial split systems via rational domain separation. Both approaches are direct applications of foundational research into the structural and functional independence of Cas9 domains—the REC lobe for recognition and the NUC lobe for cleavage.
Compact Cas9 Orthologs: Mining Nature's Toolkit
Natural evolution has produced a diversity of Cas9 proteins with varying sizes. Identifying orthologs smaller than SpCas9 provides direct solutions for viral delivery.
Key Compact Orthologs & Quantitative Comparison
The table below summarizes the characteristics of leading compact Cas9 orthologs.
Table 1: Comparison of Compact Cas9 Orthologs
| Ortholog | Source Organism | Size (aa) | PAM Sequence | Editing Efficiency (Relative to SpCas9) | Key Advantage | Primary Limitation |
|---|---|---|---|---|---|---|
| SaCas9 | Staphylococcus aureus | 1053 | 5'-NNGRRT-3' | ~50-70% | Fits in AAV with extensive regulatory elements. | Limited PAM availability. |
| CjCas9 | Campylobacter jejuni | 984 | 5'-NNNNRYAC-3' | ~30-50% | Very small; good for dual-vector AAV delivery. | Lower efficiency; complex PAM. |
| Nme2Cas9 | Neisseria meningitidis | 1082 | 5'-NNNNCC-3' | ~40-80% | High specificity; good balance of size & activity. | PAM less stringent but longer. |
| SauriCas9 | Staphylococcus auricularis | 1050 | 5'-NNGG-3' | ~60-80% | Simple NGG-like PAM; highly active. | Newer, less characterized. |
Experimental Protocol: Validating a Novel Compact Ortholog
Aim: To clone, express, and assess the genome-editing activity of a newly identified compact Cas9 ortholog in mammalian cells.
Materials & Methods:
- Gene Synthesis & Cloning: Codon-optimize the Cas9 ortholog gene for mammalian expression. Clone into a mammalian expression plasmid (e.g., pX backbone) downstream of a constitutive promoter (e.g., CMV).
- sgRNA Cloning: Clone a target-specific sgRNA sequence into a companion plasmid or the same plasmid using a U6 promoter.
- Cell Transfection: Co-transfect HEK293T cells (in a 24-well plate) with 500 ng of Cas9 plasmid and 250 ng of sgRNA plasmid using a standard transfection reagent (e.g., PEI or Lipofectamine 3000).
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract genomic DNA using a commercial kit.
- Editing Efficiency Analysis (T7 Endonuclease I Assay):
- PCR-amplify the target genomic region (amplicon size: 400-600 bp).
- Hybridize: Denature and re-anneal the PCR product to form heteroduplex DNA in the presence of mismatches from editing.
- Digest: Incubate with T7E1 enzyme, which cleaves mismatched DNA.
- Analysis: Run the digested product on an agarose gel. Quantify the band intensities of cleaved and uncut products.
- Efficiency Calculation: % Indel = 100 × (1 - sqrt(1 - (b+c)/(a+b+c))), where a is the intensity of the uncut band, and b & c are the intensities of the cut bands.
- Next-Generation Sequencing (NGS) Validation: For high-fidelity analysis, perform targeted amplicon sequencing of the PCR products and analyze indels using bioinformatics tools (e.g., CRISPResso2).
Split-Cas9 Systems: Engineering Deliverability via Domain Separation
When naturally compact orthologs are unsuitable (due to PAM or specificity), SpCas9 can be split into two or more fragments that reconstitute activity upon delivery. This strategy is predicated on the structural separation between the REC and NUC lobes.
Principles of Splitting
The split site is chosen in a surface-exposed, flexible loop connecting two structurally independent domains. Common split sites for SpCas9 are between residues 573/574 (intradomain split within REC lobe) or 713/714 (interdomain split between REC and NUC lobes). The fragments are typically fused to protein-protein interaction domains (e.g., FKBP/FRB, inteins) or self-associating peptides to facilitate reconstitution.
Diagram 1: Conceptual Basis for Cas9 Splitting
Experimental Protocol: Implementing a Dimerizer-Dependent Split-Cas9 System
Aim: To create a chemically inducible split-SpCas9 system and measure its on-target editing efficiency relative to wild-type.
Materials & Methods:
- Plasmid Construction:
- Split Cas9 at a chosen site (e.g., 713/714).
- Fuse the N-terminal fragment (Cas9-N) to FKBP12 (F36V).
- Fuse the C-terminal fragment (Cas9-C) to FRB.
- Clone each fragment into separate AAV ITR-containing plasmids with appropriate promoters.
- Virus Production & Delivery: Produce AAV serotype 9 (AAV9) vectors for each fragment. Co-inject mice (e.g., tail vein) with equal titers (e.g., 5e10 vg each) of both AAVs. Include a third AAV encoding the target sgRNA.
- Induction: Administer the dimerizer drug Rapalog (or analogous compound) intraperitoneally to induce FKBP-FRB interaction and Cas9 fragment reconstitution.
- Efficiency & Specificity Assessment:
- Harvest target tissues (e.g., liver) 2-4 weeks post-injection.
- Extract genomic DNA and analyze on-target editing at the intended locus via NGS (as in Section 2.2).
- Assess off-target effects by performing GUIDE-seq or CIRCLE-seq on treated samples.
Table 2: Performance Metrics of Representative Split-Cas9 Systems
| Split System Type | Split Site (SpCas9) | Reconstitution Method | On-Target Efficiency (% of WT) | Background Activity (No Induction) | Key Application |
|---|---|---|---|---|---|
| Intein-Mediated | 573/574 | Protein Splicing | 20-40% | Low | Single AAV delivery of dual fragments. |
| Dimerizer-Inducible | 713/714 | FKBP/FRB + Rapalog | 50-80% | Very Low (<1%) | Temporally controlled in vivo editing. |
| Direct Fusion (N/C) | 713/714 | High-affinity peptides | 10-30% | High | Proof-of-concept for reconstitution. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Compact & Split-Cas9 Research
| Item Name | Function/Benefit | Example Vendor/Product |
|---|---|---|
| AAVpro Helper Free System | Produces high-titer, pure AAV for fragment delivery in vivo. | Takara Bio |
| Lipofectamine CRISPRMAX | Optimized lipid nanoparticle for co-transfection of Cas9/sgRNA plasmids in vitro. | Thermo Fisher Scientific |
| T7 Endonuclease I | Detects indel mutations via mismatch cleavage; cost-effective for initial screening. | New England Biolabs |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR for amplifying genomic target regions prior to editing analysis. | Roche |
| CRISPResso2 Analysis Tool | Open-source software for precise quantification of genome editing from NGS data. | PMID: 30661751 |
| Alt-R S.p. HiFi Cas9 Nuclease | High-fidelity wild-type SpCas9 control for benchmarking ortholog/split system activity. | Integrated DNA Technologies |
| Dimerizer Reagents (e.g., AP21967) | Small molecule inducers for controlled protein dimerization in split systems. | Takara Bio (Clontech) |
| HEK293T/HEK293 Cells | Standard, easily transfected mammalian cell line for initial functional validation. | ATCC |
Diagram 2: Workflow for Developing a Split-Cas9 Therapeutic
The challenges of delivering the CRISPR-Cas9 machinery are being surmounted through applied structural biology. The strategies of deploying compact orthologs and engineered split systems are not merely workarounds but are direct implementations of the core thesis that Cas9 is a modular protein composed of functionally separable domains. The future of in vivo therapeutic genome editing lies in the continued refinement of these architectures—engineering smaller, more precise, and conditionally active Cas9 variants—guided by an ever-deeper understanding of the protein's structural organization.
Cas9 Structural Variants in Review: Validating Orthologs and Engineered Proteins for Specific Applications
This whitepaper provides an in-depth structural and functional analysis of Streptococcus pyogenes Cas9 (SpCas9), contextualized within a broader thesis on Cas9 protein domain architecture. As the pioneering and most characterized CRISPR-associated nuclease, SpCas9 serves as the archetype for understanding structure-function relationships in programmable genome editing.
Structural Organization of SpCas9
SpCas9 is a multi-domain, bilobed protein (~160 kDa) comprising a Recognition (REC) lobe and a Nuclease (NUC) lobe. The protein functions as a monomer, with key domains coordinating target DNA interrogation and cleavage.
Table 1: Quantitative Summary of SpCas9 Structural Domains
| Domain/Lobe | Amino Acid Residues (Approx.) | Primary Function | Key Structural Motifs |
|---|---|---|---|
| REC Lobe | 1-713 | sgRNA & DNA target recognition/binding, conformational activation. | REC1, REC2, REC3, Bridge Helix (BH). |
| NUC Lobe | 714-1368 | DNA cleavage & protospacer adjacent motif (PAM) interaction. | PAM-Interacting (PI), HNH, RuvC. |
| HNH Nuclease Domain | 775-908 | Cleaves the complementary (target) DNA strand. | ββα-metal fold. |
| RuvC-like Nuclease Domain | 1-59, 718-769, 909-1093 | Cleaves the non-complementary (non-target) DNA strand. | RNase H fold (split into 3 subdomains). |
| PI Domain | 1094-1368 | Recognizes the 5'-NGG-3' PAM sequence on dsDNA. | α-helical, PAM-reading loops. |
Functional Mechanism & Catalytic Cycle
The catalytic cycle involves a sequence of orchestrated conformational changes triggered by PAM binding and RNA-DNA heteroduplex formation.
Diagram 1: SpCas9 DNA Targeting & Cleavage Cascade
Key Experimental Protocols for Structural & Functional Analysis
Protocol 1: Cryo-EM Structure Determination of SpCas9:Target DNA Complex
- Objective: Determine high-resolution 3D structure of catalytically active or inactive (dCas9) SpCas9 bound to sgRNA and target DNA.
- Methodology:
- Protein Purification: Recombinant His-tagged SpCas9 expressed in E. coli and purified via Ni-NTA affinity, ion-exchange, and size-exclusion chromatography (SEC).
- Complex Formation: Incubate purified SpCas9 with in vitro transcribed sgRNA (1:1.2 molar ratio) for 15 min at 25°C. Add target dsDNA oligonucleotide containing a 5'-NGG-3' PAM (1:1.5 molar ratio) and incubate further.
- Vitrification: Apply 3.5 µL of complex (2.5 mg/mL) to glow-discharged Quantifoil grids, blot, and plunge-freeze in liquid ethane.
- Data Collection & Processing: Acquire movies on a 300 keV cryo-electron microscope (e.g., Titan Krios). Use motion correction, CTF estimation, particle picking (e.g., cryoSPARC), 2D classification, ab-initio reconstruction, and 3D refinement to generate a density map.
- Model Building & Refinement: Fit existing crystal structures (e.g., PDB: 4OO8) into the map using Chimera, followed by iterative manual rebuilding in Coot and refinement in Phenix.
Protocol 2: Single-Molecule FRET (smFRET) to Monitor Conformational Dynamics
- Objective: Real-time observation of DNA bending, strand separation, and lobe rearrangement during R-loop formation.
- Methodology:
- Labeling: Site-specifically label SpCas9 (e.g., via engineered cysteines in REC and NUC lobes) with donor (Cy3) and acceptor (Cy5) fluorophores. Alternatively, label dsDNA ends with FRET pair.
- Surface Immobilization: Biotinylate DNA substrates and immobilize on a PEG-passivated, streptavidin-coated quartz slide within a microfluidic flow chamber.
- Data Acquisition: Introduce labeled SpCas9-sgRNA complex and image using a total internal reflection fluorescence (TIRF) microscope. Excite donor with a 532 nm laser and collect emission from donor and acceptor channels.
- Analysis: Calculate FRET efficiency (E) for individual molecules over time. Identify distinct FRET states corresponding to "unbound," "PAM-bound," "partially unwound," and "fully formed R-loop" conformations.
Protocol 3: In Vitro Cleavage Assay for Kinetic Analysis
- Objective: Quantify nuclease activity and measure kinetic parameters (kcat, Km).
- Methodology:
- Reaction Setup: Combine 100 nM SpCas9:sgRNA ribonucleoprotein (RNP) with varying concentrations (10-500 nM) of target DNA substrate in reaction buffer (20 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol).
- Time-Course Reaction: Initiate cleavage by transferring mixture to 37°C. Aliquot at time points (e.g., 0, 15s, 30s, 1, 2, 5, 10, 30 min) and quench with 2x stop buffer (95% formamide, 20 mM EDTA).
- Product Analysis: Denature samples, resolve products via urea-PAGE (15-20%), and visualize with SYBR Gold staining.
- Quantification: Use densitometry to calculate fraction cleaved. Plot initial velocity vs. substrate concentration and fit data to the Michaelis-Menten equation to derive kcat and Km.
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function in SpCas9 Research | Example/Notes |
|---|---|---|
| Recombinant SpCas9 Protein | Core nuclease for in vitro biochemical, structural, and cleavage assays. | Commercial sources (e.g., NEB, Thermo) or in-house expression (pET-based vectors). |
| sgRNA (Synthetic or IVT) | Guides SpCas9 to specific DNA target sequence. | Chemically synthesized crRNA+tracrRNA or single-guide RNA (sgRNA) via T7 transcription. |
| PAM-containing DNA Substrates | Target for cleavage, binding, and structural studies. | Defined dsDNA oligonucleotides with varying flanking sequences for specificity analysis. |
| D10A/H840A "Dead" Cas9 (dCas9) | Catalytically inactive mutant for structural studies, imaging, or transcriptional modulation without cleavage. | Base for fusion proteins (e.g., transcriptional activators, base editors). |
| Cryo-EM Grids (Quantifoil) | Support film for vitrified sample in cryo-electron microscopy. | Au or Cu grids with 1.2/1.3 µm hole size and hole spacing. |
| Fluorophores for smFRET | Donor/Acceptor pair for monitoring nanometer-scale distance changes. | Cy3/Cy5 or Alexa Fluor 555/647, attached via maleimide (cysteine) or NHS ester (amine) chemistry. |
| Ni-NTA Resin | Affinity purification of polyhistidine-tagged SpCas9. | Critical first step in protein purification workflow. |
| Size-Exclusion Chromatography (SEC) Column | Final polishing step to isolate monodisperse, properly folded SpCas9. | e.g., Superdex 200 Increase, for analysis of complex assembly. |
Diagram 2: Core SpCas9 Domain Functional Integration
Quantitative Functional Parameters
Table 2: Key Biochemical and Biophysical Parameters of Wild-Type SpCas9
| Parameter | Measured Value | Experimental Context / Notes |
|---|---|---|
| Molecular Weight | ~158 kDa (1368 aa) | Calculated from amino acid sequence. |
| PAM Specificity | 5'-NGG-3' (canonical) | In vivo and in vitro consensus; NAG recognized with lower efficiency. |
| DNA Cleavage Rate (k_cat) | ~0.5 - 5 s⁻¹ | Varies with substrate sequence and reaction conditions (Mg²⁺, temp). |
| Dissociation Constant (K_d) for DNA | Low pM - nM range | For fully complementary target post R-loop formation. |
| R-loop Formation Kinetics | ~10-50 ms (base pairing step) | Measured via smFRET; PAM binding is rate-limiting. |
| DSB Product | Blunt ends, 5' phosphate, 3' hydroxyl | Cleavage occurs 3 bp upstream of PAM. |
Within the broader research on Cas9 protein domain architecture and structural organization, the discovery of compact Cas9 orthologs has been transformative for applications with strict size limitations, such as adeno-associated virus (AAV) delivery for gene therapy. Staphylococcus aureus Cas9 (SaCas9) emerged as a critical alternative to the commonly used Streptococcus pyogenes Cas9 (SpCas9) due to its significantly smaller size while retaining robust DNA-cleaving activity. This technical guide provides an in-depth structural comparison of SaCas9 with other small orthologs, framing their distinct architectures within the functional constraints of genome editing.
Quantitative Structural Comparison of Compact Cas9 Orthologs
Table 1: Core Quantitative Parameters of Small Cas9 Orthologs
| Ortholog (Species) | Protein Size (aa) | PAM Sequence (5'→3')* | Structural Domains | RuvC Active Site Motif | HNH Active Site Motif | Reported Editing Efficiency (%) |
|---|---|---|---|---|---|---|
| S. aureus (SaCas9) | 1053 | NNGRRT (or NNGRR) | REC I, REC II, Bridge Helix, PAM-Interacting (PI), RuvC, HNH | D10, E477, D571 (Sa) | N580, H557, D651 (Sa) | 10-50 (mammalian cells) |
| C. jejuni (CjCas9) | 984 | NNNNRYAC | REC, Bridge Helix, PI, RuvC, HNH | D8, E400, D572 (Cj) | N563, H540, D637 (Cj) | 5-40 (mammalian cells) |
| N. meningitidis (NmCas9) | 1082 | NNNNGATT | REC, Bridge Helix, PI, RuvC, HNH, Topo Homology | D16, E466, D563 (Nm) | H557, N572, D640 (Nm) | 20-60 (mammalian cells) |
| S. thermophilus (St1Cas9) | 1121 | NNAGAAW | REC I, REC II, Bridge Helix, PI, RuvC, HNH, WED | D10, E478, D571 (St1) | N580, H557, D651 (St1) | 15-45 (bacterial models) |
PAM: Protospacer Adjacent Motif. *Efficiency is highly dependent on target locus and delivery method; values represent common ranges reported in literature.
Table 2: Crystallographic and Biophysical Data
| Ortholog | PDB ID (Example) | Resolution (Å) | Overall Architecture Comparison to SpCas9 | Key Structural Distinction |
|---|---|---|---|---|
| SaCas9 | 5CZZ | 2.7 Å | ~1,000 aa smaller; similar bilobed (REC-nuclease) architecture | Shorter REC lobe; unique PI domain conformation for NNGRRT PAM recognition. |
| CjCas9 | 5X2H | 2.8 Å | Most compact; significantly truncated REC lobe. | Minimal REC domain; requires a longer PAM (8 bp), impacting target range. |
| NmCas9 | 4UNO | 2.2 Å | Similar size to SaCas9; distinct Topo homology domain insertion. | Presence of a Topoisomerase homology domain of unknown function in the HNH domain insertion. |
| St1Cas9 | 5H32 | 2.5 Å | Larger than SaCas9; contains an additional WED domain. | WED domain contributes to PAM recognition specificity for its unique PAM. |
Detailed Experimental Protocols for Structural Analysis
Protocol: Protein Purification for Crystallography/NMR (SaCas9 Example)
Objective: Obtain high-purity, monodisperse SaCas9 protein for structural studies.
- Cloning: Codon-optimize saCas9 gene for expression in E. coli BL21(DE3). Clone into a pET-based vector with an N-terminal 6xHis-SUMO or 6xHis-GST tag for improved solubility.
- Expression: Transform plasmid into expression cells. Grow culture in LB at 37°C to OD600 ~0.6. Induce with 0.5 mM IPTG and shift temperature to 18°C for 16-20 hours.
- Lysis: Harvest cells via centrifugation. Resuspend pellet in Lysis Buffer (50 mM Tris pH 8.0, 500 mM NaCl, 10% glycerol, 5 mM imidazole, 1 mM TCEP, protease inhibitors). Lyse via sonication or homogenization.
- Purification: Clarify lysate by ultracentrifugation. Load supernatant onto a Ni-NTA affinity column. Wash with 10 column volumes (CV) of Wash Buffer (Lysis Buffer with 30 mM imidazole). Elute with Elution Buffer (Lysis Buffer with 300 mM imidazole).
- Tag Cleavage & Reverse Affinity: Add SUMO or GST protease (e.g., Ulp1, TEV) and dialyze overnight at 4°C. Pass sample back over Ni-NTA or glutathione resin to remove cleaved tag and protease.
- Size Exclusion Chromatography (SEC): Concentrate protein and inject onto a pre-equilibrated HiLoad 16/600 Superdex 200 pg column in SEC Buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM TCEP). Collect the monodisperse peak.
- Quality Control: Assess purity via SDS-PAGE (>95%). Confirm homogeneity via analytical SEC or dynamic light scattering (DLS). Concentrate to 5-10 mg/mL for crystallization trials or NMR.
Protocol: Cryo-EM Sample Preparation for DNA-Bound Complex
Objective: Visualize the ternary complex (SaCas9:sgRNA:target DNA) for mechanistic insight.
- Complex Formation: Incubate purified SaCas9 with a 1.2x molar excess of chemically synthesized sgRNA (crRNA:tracrRNA duplex) for 10 min at 25°C. Add a 1.5x molar excess of target DNA duplex containing the correct PAM. Incubate 15 min.
- Grid Preparation: Apply 3 µL of complex (~0.5-1 mg/mL) to a glow-discharged (plasma cleaner) Quantifoil R 1.2/1.3 Au 300 mesh grid.
- Vitrification: Blot for 3-5 seconds at 100% humidity, 4°C, and plunge-freeze in liquid ethane using a Vitrobot (FEI).
- Data Collection: Screen grids on a 300 keV cryo-TEM (e.g., Titan Krios). Collect ~3,000 micrograph movies at a defocus range of -1.5 to -2.5 µm.
- Processing: Motion correction and CTF estimation (MotionCor2, Gctf). Automated particle picking (e.g., crYOLO). 2D classification, 3D initial model generation, and high-resolution refinement (Relion, cryoSPARC).
- Model Building: Fit the existing crystal structure (PDB: 5CZZ) into the EM map using Chimera. Manually adjust and rebuild regions (especially flexible REC lobe and nucleic acids) in Coot. Refine with Phenix.
Visualization of Key Concepts
Diagram 1: The rationale for exploring compact Cas9 orthologs.
Diagram 2: SaCas9 domain organization and functional interactions.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Compact Cas9 Research
| Reagent/Material | Function/Description | Example Vendor/Product |
|---|---|---|
| Expression Vectors | Codon-optimized plasmids for high-yield protein expression in E. coli or mammalian cells. | Addgene: pET28b-SaCas9, pX601-AAV-CBh-SaCas9 (for in vivo). |
| Purification Resins | Affinity matrices for tag-based purification (His-tag, GST-tag). | Cytiva: HisTrap HP, GSTrap HP. |
| Size Exclusion Columns | High-resolution SEC for polishing and complex analysis. | Cytiva: HiLoad Superdex 200 pg. |
| Synthetic sgRNA & DNA Oligos | Chemically synthesized, high-purity nucleic acids for complex formation and assays. | IDT: Alt-R CRISPR-Cas9 sgRNA, target DNA duplexes. |
| Cryo-EM Grids | Specimen support films for vitrification. | EMS: Quantifoil R 1.2/1.3 Au 300 mesh. |
| Crystallization Screens | Sparse matrix screens for identifying initial crystallization conditions. | Molecular Dimensions: Morpheus, JC SG. |
| Cell Lines for Functional Assays | Reporter cell lines (e.g., GFP disruption) to test editing efficiency. | ATCC: HEK293T, U2OS. |
| In Vivo Delivery Vectors | AAV vectors (e.g., AAV9, AAV-DJ) for packaging and delivering compact Cas9 in vivo. | Vigene Biosciences: AAV serotype kits. |
| Next-Gen Sequencing Kits | For deep sequencing of target loci to quantify editing outcomes and specificity. | Illumina: MiSeq Reagent Kit v3. |
This whitepaper explores the critical relationship between engineered structural modifications in high-fidelity Cas9 variants and their resultant biochemical specificity and on-target efficacy. This analysis is situated within the broader thesis that the domain architecture and structural organization of the Cas9 endonuclease—comprising the REC (Recognition), NUC (Nuclease), and PAM-interacting lobes—are not merely static scaffolds but dynamically integrated systems. Targeted perturbations within this architecture, aimed at reducing non-target DNA interactions, can have profound and sometimes unpredictable consequences for catalytic efficiency and DNA recognition fidelity. The drive to decouple specificity from activity presents a central challenge in therapeutic genome editing.
Structural Basis of High-Fidelity Engineering
The canonical Streptococcus pyogenes Cas9 (SpCas9) engages target DNA through a conformational transition from an inactive to an active state, facilitated by DNA complementarity and PAM recognition. High-fidelity variants (e.g., SpCas9-HF1, eSpCas9(1.1), HypaCas9, SpCas9-NG) introduce strategic mutations, primarily within the REC3 domain and the positively charged groove bridging the REC and NUC lobes. These mutations are designed to destabilize non-canonical DNA interactions without affecting optimal, on-target binding and catalysis.
Table 1: Key High-Fidelity Cas9 Variants and Their Structural Modifications
| Variant Name | Primary Structural Locus | Key Amino Acid Substitutions (SpCas9 Numbering) | Proposed Structural Mechanism |
|---|---|---|---|
| SpCas9-HF1 | REC3 / DNA Interface | N497A, R661A, Q695A, Q926A | Reduces non-specific electrostatic interactions with the DNA phosphate backbone. |
| eSpCas9(1.1) | Positively Charged Groove | K848A, K1003A, R1060A | Alleviates excessive stability of DNA duplex binding, particularly for off-targets. |
| HypaCas9 | REC3 & HNH Domain | N692A, M694A, Q695A, H698A | Stabilizes the HNH nuclease domain in an inactive conformation until correct proofreading. |
| xCas9 3.7 | REC2, REC3, PI | E121A, D133A, R324A, T327A, E409A, etc. | Broadens PAM recognition (NG, GAA) while increasing fidelity via multiple domain tweaks. |
| SpCas9-NG | PAM-Interacting Domain | R1335V/L, L1111R, D1135V, G1218R, etc. | Alters PAM specificity to NG; fidelity is a secondary characteristic of altered PAM interrogation. |
Impact on Specificity: Quantitative Analysis
Specificity is quantified using genome-wide methods such as GUIDE-seq, CIRCLE-seq, BLISS, or Digenome-seq. These techniques identify off-target sites with indel frequencies, allowing for the calculation of specificity indices.
Table 2: Comparative Specificity Profiles of Selected HiFi Variants
| Variant | Average Reduction in Off-Target Activity (vs. wtSpCas9) | Method | Notable Trade-off Observed |
|---|---|---|---|
| SpCas9-HF1 | >85% across 12 known off-targets | GUIDE-seq | Significant on-target reduction at some loci (up to 70%). |
| eSpCas9(1.1) | ~90% reduction | BLISS | Less pronounced on-target reduction than HF1, but context-dependent. |
| HypaCas9 | >94% reduction at validated sites | Digenome-seq | Maintains robust on-target activity; improved proofreading. |
| Sniper-Cas9 | ~78% reduction | GUIDE-seq | Engineered for balance; often shows higher on-target than HF1/eSpCas9. |
| evoCas9 | Undetectable at most off-targets | CIRCLE-seq | Directed evolution product; maintains high on-target across diverse loci. |
Impact on On-Target Activity: Mechanistic Consequences
Structural changes that increase specificity often do so by raising the energy barrier for DNA cleavage. This can inadvertently impact on-target kinetics. Key experimental metrics include indel efficiency (%), in vitro cleavage kinetics (k~cat~, K~m~), and cellular expression/stability assays.
Table 3: Quantitative On-Target Activity Metrics
| Variant | Median On-Target Indel Efficiency (Human Cells, %)* | Relative In Vitro Cleavage Rate (k~cat~) | Cellular Abundance (Relative to wt) |
|---|---|---|---|
| wtSpCas9 | 40.5 | 1.00 | 1.00 |
| SpCas9-HF1 | 28.7 | 0.15 - 0.30 | ~0.95 |
| eSpCas9(1.1) | 33.2 | 0.25 - 0.40 | ~1.02 |
| HypaCas9 | 38.9 | ~0.70 | ~0.90 |
| evoCas9 | 39.5 | ~0.85 | ~1.10 |
*Data synthesized from multiple studies (2016-2023) across 20+ genomic loci.
Detailed Experimental Protocols
Protocol:In VitroCleavage Kinetics Assay (Stopped-Flow Fluorescence)
Purpose: Measure the catalytic rate constant (k~cat~) and Michaelis constant (K~m~) for Cas9 ribonucleoprotein (RNP) complexes. Reagents: Purified Cas9 protein, synthetic sgRNA, dual-fluorophore labeled DNA substrate (FAM donor, TAMRA acceptor). Procedure:
- RNP Formation: Pre-complex purified Cas9 (100 nM) with sgRNA (120 nM) in cleavage buffer (20 mM HEPES pH 7.5, 150 mM KCl, 10 mM MgCl2, 5% glycerol) for 10 min at 37°C.
- Substrate Preparation: Serially dilute fluorescent target DNA (0-500 nM) in the same buffer.
- Kinetic Measurement: Load RNP and substrate solutions into a stopped-flow spectrometer. Rapidly mix equal volumes and monitor fluorescence resonance energy transfer (FRET) decrease (excitation 495 nm, emission 585 nm) over 60 seconds.
- Data Analysis: Fit the initial velocity data to the Michaelis-Menten equation (v = (V~max~[S])/(K~m~+[S])) to derive k~cat~ and K~m~.
Protocol: Genome-Wide Off-Target Detection (CIRCLE-seq)
Purpose: Identify potential off-target sites biochemically with high sensitivity. Reagents: Genomic DNA, Cas9 RNP, Circligase, Phi29 polymerase, NEXTflex barcoded adapters. Procedure:
- Genomic DNA Isolation & Shearing: Extract genomic DNA and fragment to ~300 bp via sonication.
- In Vitro Digestion: Incubate sheared DNA (1 µg) with pre-formed RNP (200 nM) for 16h at 37°C.
- Circularization: Repair ends and circularize cleaved fragments using Circligase. Linear, uncut DNA is not efficiently circularized.
- Digestion of Linear DNA: Treat with exonuclease to digest all non-circularized DNA.
- Rolling Circle Amplification: Use Phi29 polymerase to amplify circularized off-target fragments.
- Sequencing Library Prep: Fragment amplified product, ligate adapters, and sequence on an Illumina platform.
- Bioinformatics: Map sequences to reference genome, identifying junctions indicative of Cas9 cleavage sites.
Visualizations: Pathways and Workflows
Diagram Title: Cas9 Activation Pathway and HiFi Checkpoints
Diagram Title: CIRCLE-seq Off-Target Detection Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Evaluation | Key Considerations |
|---|---|---|
| Recombinant HiFi-Cas9 Proteins | Core enzyme for in vitro and cellular assays. | Purity (>95%), endotoxin levels, storage buffer composition. |
| Chemically Modified sgRNAs | Guides with 2'-O-methyl, phosphorothioate modifications. | Enhance RNP stability, reduce immune response, improve editing efficiency. |
| Synthetic Target DNA Duplexes | Fluorescently/quencher-labeled substrates for kinetic assays. | Label position (donor/acceptor pairs), purity, annealing protocol. |
| Cellular Delivery Reagents | Lipofectamine, electroporation kits (e.g., Neon). | Optimization required for each cell type and Cas9 variant RNP. |
| NGS-Based Off-Target Kits | Commercial GUIDE-seq or CIRCLE-seq kits. | Standardization, sensitivity, and background reduction. |
| Anti-Cas9 Monoclonal Antibodies | For Western blot, ELISA, or cellular localization. | Specificity for engineered variants (epitope tagging may be needed). |
| Positive Control gRNA/DNA Plasmids | Validated active and off-target sequences. | Essential for benchmarking variant performance. |
| dCas9-Based Reporter Cell Lines | For specificity screening via transcriptional activation/repression. | Provides a rapid, functional readout of DNA binding fidelity. |
The evaluation of high-fidelity Cas9 variants underscores a fundamental principle of protein engineering within the Cas9 architectural thesis: modifications to enhance one property (specificity) inevitably alter the energetic landscape of the entire catalytic cycle. The most successful variants, such as HypaCas9 and evoCas9, achieve a superior balance by introducing mutations that enforce kinetic proofreading without excessively destabilizing the catalytically competent conformation. Future engineering efforts must continue to leverage high-resolution structural data and directed evolution, focusing on the allosteric networks connecting the REC, NUC, and PAM-interacting domains to achieve the ultimate goal of a "perfect" editor—one with undetectable off-target activity and unwavering on-target potency.
The canonical Streptococcus pyogenes Cas9 (SpCas9) is defined by a multi-domain architecture that dictates its function: the REC lobe (RecI-III domains) for nucleic acid binding and conformational activation, and the NUC lobe (HNH, RuvC, and PAM-interacting domains). The PAM-interacting (PI) domain is a critical structural determinant of target range, recognizing the canonical 5'-NGG-3' sequence. Research into altering PAM specificity is fundamentally a study of PI domain engineering and its allosteric communication with the catalytic HNH and RuvC domains. The development of xCas9 and SpCas9-NG represents successful rational and directed evolution approaches to modify this architecture, broadening the targetable genomic space for research and therapeutic applications.
Engineered Cas9 Variants: Core Properties & Performance Data
A comparative summary of key biochemical and functional properties.
Table 1: Comparative Analysis of SpCas9, xCas9, and SpCas9-NG
| Property | Wild-Type SpCas9 | xCas9 (v3.7) | SpCas9-NG |
|---|---|---|---|
| Primary PAM Specificity | 5'-NGG-3' (requires G at positions 2 & 3) | 5'-NG-3' (G required only at pos. 2) | 5'-NG-3' (G required only at pos. 2) |
| Recognized PAMs | NGG (strict) | NG, GAA, GAT (relaxed) | NG (NGA, NGC, NGT, NGG) |
| Average Editing Efficiency at NG PAMs | <5% | ~30-60% (varies by site) | ~10-40% (varies by site) |
| Average Editing Efficiency at NGG PAMs | ~40-70% | Comparable or slightly reduced vs. WT | Comparable or slightly reduced vs. WT |
| Primary Engineering Method | N/A | Phage-assisted continuous evolution (PACE) | Structure-guided rational design |
| Key Mutations | N/A | A262T, R324L, S409I, E480K, E543D, M694I, E1219V | R1335V/L1111R/N1317R |
| On-Target Specificity | Standard | Increased (higher fidelity) | Comparable to WT or slightly improved |
| Size (aa) | 1368 | 1368 | 1368 |
Detailed Experimental Protocols
Protocol 1: In Vitro PAM Depletion Assay (Determining Novel PAM Specificity)
Purpose: To comprehensively identify DNA sequences recognized as functional PAMs by an engineered Cas9 variant. Reagents:
- Purified Cas9 protein (WT and engineered variant).
- In vitro-transcribed sgRNA targeting a neutral sequence.
- Plasmid library containing a randomized 8-bp PAM region (5'-NNNNNNNN-3') upstream of the target protospacer.
- NEBuffer r3.1, dNTPs.
- Primers for amplifying the randomized region for sequencing.
- High-fidelity DNA polymerase, NEXTflex barcodes for Illumina sequencing.
Procedure:
- Form RNP Complexes: Incubate 100 nM Cas9 protein with 120 nM sgRNA in reaction buffer for 10 min at 25°C.
- Digestion Reaction: Add 10 ng of the plasmid library to the RNP mix. Incubate at 37°C for 1 hour to allow for PAM recognition, DNA cleavage, and plasmid linearization.
- Depletion of Cleaved Plasmids: Treat the reaction with Plasmid-Safe ATP-dependent DNase for 2 hours at 37°C. This enzyme degrades linearized DNA, enriching for circular (uncleaved) plasmids.
- Amplification & Sequencing: PCR-amplify the randomized PAM region from the uncleaved plasmid pool using barcoded primers. Perform deep sequencing (Illumina MiSeq).
- Data Analysis: Compare the frequency of each 8-mer sequence in the pre- and post-selection libraries. Depleted sequences represent functional PAMs. Calculate an enrichment score (log2[initial/final]) for each NNN sequence.
Protocol 2: Cellular Editing Efficiency Assessment via T7 Endonuclease I (T7E1) Assay
Purpose: To quantify genome editing efficiency at endogenous loci with candidate PAMs in mammalian cells. Reagents:
- HEK293T cells.
- Expression plasmids: pX458 (or similar) encoding Cas9 variant and sgRNA.
- Lipofectamine 3000 transfection reagent.
- Genomic DNA extraction kit.
- PCR primers flanking the target locus.
- T7 Endonuclease I enzyme, NEBuffer 2.
- Agarose gel electrophoresis system.
Procedure:
- Cell Transfection: Seed HEK293T cells in 24-well plates. Transfect with 500 ng of Cas9-sgRNA plasmid per well using Lipofectamine 3000 according to manufacturer protocol.
- Genomic DNA Harvest: 72 hours post-transfection, extract genomic DNA.
- PCR Amplification: Perform PCR on the target locus (~500-800 bp amplicon) using high-fidelity polymerase.
- Heteroduplex Formation: Denature and reanneal the PCR products: 95°C for 5 min, ramp down to 85°C at -2°C/sec, then to 25°C at -0.1°C/sec.
- T7E1 Digestion: Digest the reannealed products with 0.5 µL of T7E1 enzyme at 37°C for 25 minutes.
- Analysis: Run digested products on a 2% agarose gel. Cleaved bands indicate presence of indel mutations. Calculate editing efficiency: (1 - sqrt(1 - (b+c)/(a+b+c))) * 100%, where a is the integrated intensity of the undigested band, and b & c are the digested band intensities.
Visualizing Engineering Strategies and Outcomes
Title: Engineering Pathways for Cas9 PAM Expansion
Title: Core Assays for PAM Characterization & Validation
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for PAM Specificity Studies
| Reagent / Material | Function & Purpose |
|---|---|
| PAM Depletion Plasmid Library | A plasmid pool with randomized nucleotides at the PAM position. Serves as the substrate for in vitro determination of all possible recognized PAM sequences by a Cas9 variant. |
| Phage-Assisted Continuous Evolution (PACE) System | A directed evolution platform using M13 bacteriophage to link Cas9 PAM recognition to phage survival, enabling rapid protein evolution over hundreds of generations. |
| T7 Endonuclease I (T7E1) | A mismatch-specific endonuclease that cleaves DNA heteroduplexes formed by reannealing PCR products from edited and wild-type alleles. Standard tool for quantifying indel frequencies. |
| HEK293T Cell Line | A highly transfectable, human embryonic kidney cell line. The standard workhorse for initial in cellulo validation of CRISPR-Cas9 editing efficiency and specificity. |
| pX458 (or pX459) Vector | A mammalian all-in-one expression plasmid encoding SpCas9 (or variant), a sgRNA scaffold, and a fluorescent marker (GFP)/puromycin resistance for transfection tracking/selection. |
| Next-Generation Sequencing (NGS) Library Prep Kits | For high-throughput, quantitative analysis of editing outcomes (indel spectra) and PAM depletion assay results. Provides base-pair resolution data. |
| Recombinant Cas9 Protein (WT & Engineered) | Purified protein for in vitro biochemical assays (PAM depletion, cleavage kinetics) and for forming ribonucleoprotein (RNP) complexes for delivery. |
The revolutionary potential of CRISPR-Cas9 genome editing is mediated by the multi-domain architecture of the Cas9 protein. Key domains, including the REC lobes (for guide RNA and target DNA recognition), the HNH and RuvC nuclease domains (for DNA cleavage), and the PAM-interacting (PI) domain, collectively determine the enzyme's operational parameters. Research into this structural organization reveals that natural variations and protein engineering alter critical performance metrics: size (affecting delivery), PAM (protospacer adjacent motif) requirement (defining targetable genomic loci), fidelity (specificity, minimizing off-target effects), and on-target editing efficiency. The selection of an optimal Cas9 variant or derivative is therefore a critical, context-dependent decision, differing fundamentally between therapeutic applications and basic research. This guide provides a decision matrix, grounded in the latest structural and functional data, to navigate this selection process.
Quantitative Comparison of Key Cas9 Variants and Derivatives
The following tables summarize the core characteristics of prominent Cas9-based tools, with data aggregated from recent primary literature (2023-2024).
Table 1: Core Characteristics of Primary Cas9 Orthologs and Common Derivatives
| Tool Name | Size (aa) | PAM Requirement (5'->3') | Key Fidelity Features | Typical On-Target Efficiency (in cells) | Primary Use Context |
|---|---|---|---|---|---|
| SpCas9 (S. pyogenes) | 1368 | NGG (canonical) | Standard; prone to off-targets with NGG/NAG | 20-60% (varies by locus) | Broad research workhorse |
| SpCas9-HF1 | 1368 | NGG | High-fidelity; engineered via alanine mutations to reduce non-specific contacts | 15-50% (often slightly reduced vs. WT) | Research requiring high specificity |
| SpCas9-eSpCas9(1.1) | 1368 | NGG | Enhanced specificity; mutations to reduce non-target DNA binding | 15-50% (similar to HF1) | Research requiring high specificity |
| SaCas9 (S. aureus) | 1053 | NNGRRT (or NNNRRT) | Moderate; smaller size improves AAV delivery but PAM is more restrictive | 10-40% | Therapeutic (in vivo delivery via AAV) |
| Nme2Cas9 (N. meningitidis) | 1082 | NNNNGATT | Very high; natural high fidelity due to stringent PAM and process | 10-30% | Research & potential therapeutic (high fidelity, small size) |
| Cas9 nickase (nCas9-D10A) | 1368 | NGG | Paired nicking increases fidelity by >1000-fold; requires two guides | Varies (paired nicking) | Research requiring extreme precision; base editing fusion |
| Catalytically dead Cas9 (dCas9) | 1368 | NGG | No cleavage; used for repression/activation (CRISPRi/a) | N/A (binding efficiency high) | Gene regulation research |
Table 2: Engineered & Evolved Variants with Altered PAMs
| Tool Name | Size (aa) | PAM Requirement (5'->3') | Parent | Key Feature | Potential Context |
|---|---|---|---|---|---|
| SpCas9-VQR | 1368 | NGA | SpCas9 | Engineered PI domain | Research for targeting AT-rich regions |
| SpCas9-NG | 1368 | NG | SpCas9 | Relaxed PAM (vs NGG) | Broad research, increased target range |
| xCas9 3.7 | 1368 | NG, GAA, GAT | SpCas9 | Broad PAM, improved fidelity | Research with flexible PAM needs |
| SpRY (near PAM-less) | 1368 | NRN > NYN | SpCas9 | Virtually PAM-less | Ultimate research flexibility; fidelity trade-off |
| Sc++ (S. canis) | ~1370 | NNG | ScCas9 | Evolved for broader NG PAM | Research, potential alternative to SpCas9-NG |
The Decision Matrix: Therapeutic vs. Research Prioritization
The selection is driven by ranking the four core metrics based on the application's primary constraints and goals.
For Therapeutic Development (e.g., in vivo gene therapy):
- Size: Highest Priority. Must be packageable into delivery vectors (e.g., AAV ~4.7kb limit). Variants like SaCas9 or Nme2Cas9 are preferred.
- Fidelity: Critical. Minimizing off-targets is mandatory for clinical safety. High-fidelity variants (e.g., Nme2Cas9, HypaCas9) or paired-nicking strategies are essential.
- PAM: Constraining Factor. Must be compatible with the therapeutic target locus. May require engineering or choosing a specific ortholog.
- Efficiency: Important but Secondary. Must achieve therapeutic threshold, but not at the cost of size or fidelity. Optimized via guide design and delivery.
For Basic Research (e.g., in vitro or cell line studies):
- PAM & Efficiency: Highest Priority. Maximizing targetable loci and achieving robust editing for phenotypic analysis is key. SpCas9, SpCas9-NG, or SpRY are often first choices.
- Fidelity: Context-Dependent. Critical for genotype-phenotype studies; less so for bulk cell selection screens. SpCas9-HF1/eSpCas9 or xCas9 balance fidelity and performance.
- Size: Lowest Priority (for standard transfection/electroporation). Larger proteins like SpCas9 are not a limitation.
Experimental Protocols for Key Validation Experiments
Protocol 1: Assessing On-Target Editing Efficiency (NGS-Based)
- Objective: Quantify indel formation percentage at a specific genomic locus.
- Materials: Cultured cells, Cas9/gRNA RNP or plasmid, PCR reagents, NGS library prep kit.
- Method:
- Transfection: Deliver Cas9 and target-specific sgRNA into cells.
- Harvest: After 72h, extract genomic DNA.
- Amplification: Perform PCR (~300-400bp amplicon) flanking the target site using barcoded primers.
- Library Prep & Sequencing: Purify PCR products, pool, and prepare for Illumina MiSeq (2x250bp).
- Analysis: Use CRISPResso2 or similar tool to align reads to reference and quantify % indels.
Protocol 2: Evaluating Specificity (Genome-Wide Off-Target Analysis - CIRCLE-seq)
- Objective: Identify potential off-target sites genome-wide in an in vitro setting.
- Materials: Purified Cas9 protein, sgRNA, genomic DNA, CIRCLE-seq kit components (circligase, phi29 polymerase), NGS platform.
- Method:
- Genomic DNA Preparation: Shear genomic DNA and ligate into circular molecules.
- In Vitro Cleavage: Incubate circularized DNA with Cas9:sgRNA RNP.
- Linearization of Cleaved Products: Treat with exonuclease to degrade uncircularized and uncleaved DNA. Use T7 endonuclease I or repair ligation to linearize only cleaved circles.
- Amplification & Sequencing: Amplify linearized fragments with phi29 polymerase, prepare NGS library, and sequence.
- Analysis: Map breaksites to the reference genome to identify all potential Cas9 cleavage sites.
Visualizing Selection Logic and Workflows
Diagram Title: Decision Tree for Therapeutic vs. Research Cas9 Tool Selection
Diagram Title: On-Target Efficiency NGS Workflow (76 chars)
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent / Material | Function & Rationale |
|---|---|
| Purified Recombinant Cas9 Protein | For RNP (ribonucleoprotein) delivery, offering rapid action, reduced off-targets, and no DNA integration risk. Essential for sensitive primary cells. |
| Chemically Modified Synthetic sgRNA (e.g., 2'-O-methyl 3' phosphorothioate) | Increases stability, reduces immune response, and improves editing efficiency in hard-to-transfect cells. |
| AAV Vector Serotypes (e.g., AAV9, AAV-DJ) | For in vivo delivery. Different serotypes provide varying tropism for target tissues (liver, CNS, muscle). |
| HDR Donor Template (ssODN or AAV donor) | For precise knock-ins or corrections. Single-stranded oligodeoxynucleotides (ssODNs) for short edits; AAV donors for larger inserts. |
| Next-Generation Sequencing (NGS) Kit (e.g., Illumina MiSeq) | Gold standard for unbiased quantification of on-target editing efficiency and genome-wide off-target profiling (via CIRCLE-seq, GUIDE-seq). |
| CRISPResso2 / Cas-Analyzer Software | Critical bioinformatics tools for analyzing NGS data from editing experiments to quantify indel spectra and frequencies. |
| T7 Endonuclease I (T7E1) or Surveyor Nuclease | Mismatch-specific nucleases for rapid, low-cost initial assessment of editing efficiency via gel electrophoresis. Less quantitative than NGS. |
| Validated Positive Control sgRNA & Target Plasmid | Essential experimental control to verify Cas9 protein/RNA activity. Often targets a well-characterized locus (e.g., AAVS1 safe harbor). |
| Lipofectamine CRISPRMAX or Neon Electroporation System | Optimized delivery reagents for introducing RNP or plasmid DNA into a wide range of mammalian cell types. |
Conclusion
The functional prowess of the Cas9 nuclease is inextricably linked to its elegantly organized domain architecture. Understanding the structural interplay between the REC lobe, NUC lobe, HNH, and RuvC domains is not merely an academic exercise; it is the foundational knowledge required to harness, optimize, and innovate within the CRISPR-Cas9 toolkit. From guiding precise sgRNA design to engineering next-generation variants with enhanced fidelity, relaxed PAM requirements, and compact size, every methodological and troubleshooting advance is rooted in structural insight. The comparative analysis of natural and engineered Cas9 proteins validates this approach, providing a diverse portfolio of tools tailored for specific research and clinical challenges. Future directions point toward the continued rational design of Cas9 and novel CRISPR-associated proteins, integrating cryo-EM and AI-driven structural predictions to create hyper-specific, efficient, and deliverable editors. This deep structural knowledge will be paramount in translating CRISPR technology into safe, effective, and versatile therapeutic modalities, solidifying its role in the future of biomedicine and drug development.