Maximizing Primary Cell Gene Editing: A Complete Guide to Cas9-sgRNA Complex Formation
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed exploration of Cas9 protein-sgRNA complex formation specifically for primary cell editing.
Maximizing Primary Cell Gene Editing: A Complete Guide to Cas9-sgRNA Complex Formation
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed exploration of Cas9 protein-sgRNA complex formation specifically for primary cell editing. Covering foundational principles, practical methodologies, common troubleshooting strategies, and validation techniques, this article synthesizes current best practices for achieving efficient, specific, and reproducible genome editing in primary cells—a critical but challenging biological system for therapeutic development and basic research.
Understanding the Core: The Biology and Chemistry of Cas9-sgRNA Complexes in Primary Cells
Primary cells, isolated directly from living tissue, offer unparalleled physiological relevance for disease modeling and therapeutic development. However, their use in CRISPR-Cas9 editing presents a distinct set of challenges not typically encountered with immortalized cell lines. These challenges are critically framed within the broader thesis of optimizing Cas9 protein-sgRNA (ribonucleoprotein, RNP) complex formation and delivery for primary cell research. Success hinges on overcoming barriers related to cell viability, delivery efficiency, and low proliferative rates.
Key Challenges in Primary Cell Editing
The primary obstacles stem from the very nature of primary cells: they are non-proliferative or slow-dividing, have limited ex vivo lifespans, possess intact innate immune responses, and are highly sensitive to exogenous manipulation.
Table 1: Quantitative Comparison of Editing Challenges in Primary Cells vs. Immortalized Lines
| Challenge Parameter | Primary Cells | Immortalized Cell Lines (e.g., HEK293) | Impact on Cas9-sgRNA RNP Editing |
|---|---|---|---|
| Transfection Efficiency | Typically 10-50% (method dependent) | Often >70-90% | Low delivery reduces editing pool, requiring high-activity RNP complexes. |
| Cell Division Rate | Low/Non-dividing | High, continuous | HDR editing is severely limited; favors NHEJ or alternative knock-in strategies. |
| In Vitro Lifespan | Limited (few passages) | Essentially unlimited | Narrow window for experimental manipulation and phenotypic analysis. |
| Toxicity Sensitivity | High | Relatively low | Electroporation or chemical transfection can cause high mortality. |
| Immune Response | Intact (e.g., cGAS-STING) | Often compromised | DNA transfection can trigger apoptosis; protein RNP delivery is preferred. |
| Culture Requirements | Complex, often serum-free | Standardized, robust | Adds variables that can affect RNP stability and delivery. |
Detailed Application Notes & Protocols
The following protocols are designed within the thesis framework that optimal, pre-formed Cas9-sgRNA RNP complexes offer the fastest kinetics and lowest off-target effects, which is crucial for sensitive primary cells.
Protocol 1: Formation and Validation of Cas9-sgRNA RNP Complexes for Primary Cells
Objective: To assemble and quality-check functional Cas9-sgRNA RNP complexes prior to delivery.
Materials:
- Purified recombinant S.p. Cas9 protein (commercial source)
- Chemically synthesized sgRNA (with modifications for stability, e.g., 2'-O-methyl analogs)
- Nuclease-Free Duplex Buffer (IDT) or equivalent
- Thermal cycler or heat block
Procedure:
- sgRNA Resuspension: Centrifuge sgRNA tube at 3,000 x g for 1 minute. ResusguideRNA to 100 µM in nuclease-free duplex buffer.
- Complex Assembly: In a nuclease-free microcentrifuge tube, combine:
- 3 µL of 100 µM sgRNA (300 pmol)
- 6 µL of 50 µM Cas9 protein (300 pmol)
- 11 µL of 1X PBS (Ca/Mg-free)
- Final: 20 µL volume, 15 µM RNP complex.
- Incubation: Mix gently by pipetting. Incubate at room temperature (20-25°C) for 10-20 minutes to allow complete RNP formation.
- Validation (Gel Shift Assay): Prepare a 2% agarose gel in 0.5X TBE. Mix 2 µL of RNP complex with 6X DNA loading dye (non-denaturing). Load alongside free sgRNA and Cas9 protein controls. Run at 80V for 45-60 minutes. A successful complex shows a band shift (retarded migration) compared to free sgRNA.
Protocol 2: Electroporation of Cas9-sgRNA RNP into Human T Cells
Objective: To deliver pre-formed RNP complexes into primary human T cells using a high-efficiency, low-toxicity electroporation method.
Materials:
- Isolated primary human CD3+ T cells
- Pre-formed Cas9-sgRNA RNP complex (from Protocol 1)
- P3 Primary Cell 4D-Nucleofector X Kit S (Lonza) or equivalent
- Nucleofector 4D or 2b device
- RPMI-1640 medium + 10% FBS (pre-warmed)
Procedure:
- Cell Preparation: Isolate and count CD3+ T cells. For each reaction, centrifuge 1x10^6 cells at 300 x g for 5 minutes. Aspirate supernatant completely.
- Nucleofection Sample Prep: Thaw Nucleofector Solution and Supplement. Add 20 µL Supplement to 100 µL Solution per reaction. Add 20 µL of this complete mix to the cell pellet. Gently resuspend.
- Add RNP: Add 2-5 µL of the 15 µM RNP complex (from Protocol 1) to the cell suspension. Mix gently by pipetting 2-3 times.
- Electroporation: Transfer the entire mixture (~25 µL) to a Nucleocuvette. Insert into the Nucleofector device and run the pre-optimized program for T cells (e.g., EH-115 or FF-120).
- Recovery: Immediately after pulsing, add 80 µL of pre-warmed medium to the cuvette. Using the provided pipette, gently transfer cells to a 24-well plate containing 1 mL pre-warmed medium.
- Culture: Place plate in a 37°C, 5% CO2 incubator. Analyze editing efficiency via flow cytometry (for fluorescent reporters) or genomic DNA extraction and sequencing at 48-72 hours post-electroporation.
Visualizations
Title: Primary Cell Barriers Impact CRISPR Editing Outcomes
Title: Optimized RNP Workflow for Primary Cell Editing
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Cas9-sgRNA RNP Editing in Primary Cells
| Item | Function & Rationale | Example/Brand Considerations |
|---|---|---|
| Recombinant Cas9 Protein | High-purity, endotoxin-free protein for RNP assembly. Avoids DNA vector toxicity and offers fast editing kinetics. | Alt-R S.p. Cas9 Nuclease V3 (IDT), TrueCut Cas9 Protein v2 (Thermo Fisher) |
| Chemically Modified sgRNA | Synthetic sgRNAs with 2'-O-methyl/phosphorothioate modifications increase nuclease resistance and RNP stability in vivo. Critical for primary cell efficiency. | Alt-R CRISPR-Cas9 sgRNA (IDT), Synthego sgRNA EZ Kit |
| Primary Cell Nucleofector Kit | Optimized buffers and protocols for specific primary cell types (T cells, HSCs, neurons). Maximizes viability and delivery. | P3/P4 Primary Cell Kits (Lonza), Neon Transfection System (Thermo Fisher) |
| Cell-Type Specific Media | Serum-free or specialized media maintains cell health and potency post-electroporation, supporting edited cell survival. | TexMACS (Miltenyi), StemSpan (StemCell Tech), X-VIVO (Lonza) |
| NGS-based Editing Analysis | Quantitative, unbiased measurement of indels and HDR efficiency in a heterogeneous primary cell population. | Illumina MiSeq, amplicon sequencing assays. |
| Viability & Apoptosis Assays | To quantify the toxicity of the editing procedure (RNP + delivery). Essential for protocol optimization. | Flow cytometry with Annexin V/7-AAD, Cellometer viability stains. |
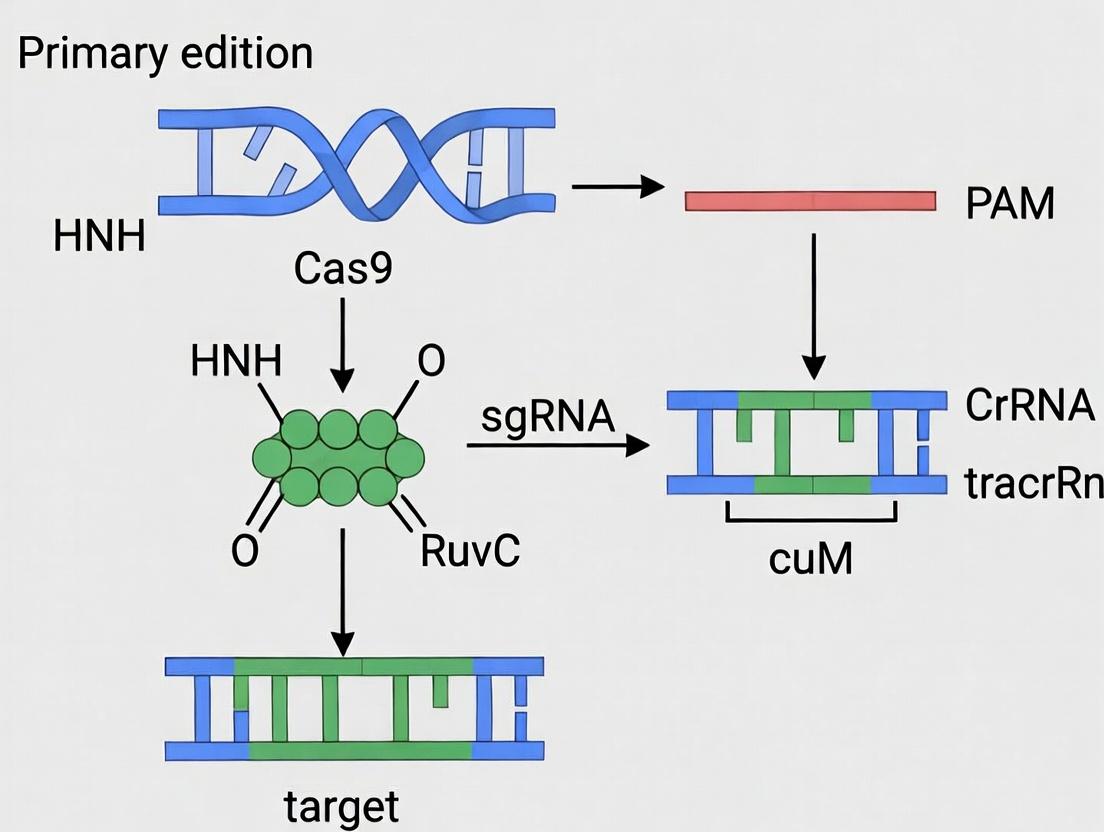
This Application Note details the molecular interactions underpinning the formation of the CRISPR-Cas9 ribonucleoprotein (RNP) complex, a critical pre-requisite for efficient genome editing, particularly in sensitive primary cell systems. Understanding these structural determinants is essential for designing high-efficiency, high-specificity RNP complexes for therapeutic development.
Key Molecular Interactions and Quantitative Data
The active complex is formed by the association of the Cas9 endonuclease with a chimeric single-guide RNA (sgRNA). The structural data reveals a bilobed architecture for Cas9 (REC and NUC lobes) that undergoes significant conformational activation upon sgRNA binding.
Table 1: Key Protein-RNA Interaction Domains and Functions
| Cas9 Domain | Interaction Target on sgRNA | Primary Function in Complex Formation |
|---|---|---|
| REC Lobe (REC1-3) | Repeat:Antirepeat Stem Loop | Facilitates sgRNA loading, mediates conformational activation for DNA binding. |
| Bridge Helix (BH) | sgRNA scaffold (phosphodiester backbone) | Stabilizes the RNA-DNA heteroduplex, contributes to cleavage activation. |
| PI Domain | Tetra-loop and stem-loop 2 | Anchors the 3' end of the sgRNA scaffold, crucial for structural integrity. |
| NTD (N-terminal Domain) | sgRNA scaffold (5' end) | Initiates binding and stabilization of the sgRNA 5' handle. |
Table 2: Thermodynamic and Kinetic Parameters of Complex Formation
| Parameter | Reported Value | Experimental Method | Implication for Editing |
|---|---|---|---|
| Dissociation Constant (Kd) | ~0.5 - 2 nM | Fluorescence Polarization (FP) / EMSA | High-affinity binding ensures stable RNP delivery. |
| Association Rate (k_on) | ~0.5 - 1 x 10^8 M^-1 s^-1 | Stopped-Flow Fluorescence | Rapid complex assembly is favorable for delivery protocols. |
| Activation Energy Barrier | Lowered upon REC lobe engagement | Single-Molecule FRET | sgRNA binding pre-orders Cas9 into a DNA-competent state. |
Detailed Protocols
Protocol 1: Fluorescence Polarization Assay for sgRNA Binding Affinity Objective: Determine the dissociation constant (Kd) of Cas9-sgRNA binding.
- Reagent Preparation: Serially dilute purified Cas9 protein (0.01 nM to 100 nM) in assay buffer (20 mM HEPES pH 7.5, 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.01% Tween-20).
- Probe Preparation: Use a 5'-fluorescein-labeled sgRNA scaffold (constant 1 nM concentration).
- Binding Reaction: Mix 50 µL of each Cas9 dilution with 50 µL of labeled sgRNA probe in a black 96-well plate. Incubate at 25°C for 30 min.
- Measurement: Read fluorescence polarization (mP units) using a plate reader (λex = 485 nm, λem = 535 nm).
- Analysis: Fit the binding isotherm (mP vs. [Cas9]) to a one-site specific binding model using non-linear regression to extract Kd.
Protocol 2: Native Gel Electrophoretic Mobility Shift Assay (EMSA) Objective: Visually confirm RNP complex formation and assess binding efficiency.
- Complex Formation: Combine 100 nM Cas9 with 50 nM sgRNA (unlabeled) in binding buffer. Incubate 15 min at 37°C.
- Gel Loading: Load samples onto a pre-run 6% native polyacrylamide gel (0.5x TBE, 2.5 mM MgCl2). Run at 100V for 45-60 min at 4°C.
- Staining: Stain the gel with SYBR Gold nucleic acid stain for 15 min.
- Imaging: Visualize using a gel imager (Ethidium Bromide channel). Unbound sgRNA migrates faster; the Cas9-sgRNA complex is retarded.
Protocol 3: RNP Complex Assembly for Primary Cell Electroporation Objective: Generate functional RNP for direct delivery into primary T cells or HSPCs.
- Component Calculation: For a 10 µL reaction, calculate amounts for a final 5 µM RNP complex. Example: 5 µL of 10 µM Cas9 protein + 5 µL of 10 µM sgRNA.
- Assembly: Combine Cas9 and sgRNA in duplex buffer (30 mM HEPES pH 7.5, 100 mM KCl). Mix gently by pipetting.
- Incubation: Incubate at room temperature for 10 minutes to allow complex formation.
- Delivery: Immediately mix the assembled RNP with cells and electroporation buffer, and proceed with nucleofection. Do not store assembled RNPs for extended periods.
Visualization of Complex Formation and Workflow
Title: Cas9-sgRNA Activation Pathway
Title: RNP Characterization & Assembly Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Structural & Functional Analysis of Cas9-sgRNA Complexes
| Reagent / Material | Function / Purpose | Example Vendor/Type |
|---|---|---|
| Recombinant Cas9 Nuclease | High-purity, endotoxin-free protein for in vitro binding assays and RNP assembly. | Commercial (e.g., IDT, Thermo), or lab-purified HiS-tagged SpCas9. |
| Chemically Modified sgRNA | Enhanced nuclease resistance and improved binding affinity for primary cell work. | 2'-O-methyl 3' phosphorothioate modifications at terminal nucleotides. |
| Fluorescently-Labeled sgRNA | Essential for FP binding assays and live-cell tracking of RNP complexes. | 5'-FAM or 5'-Cy5 labeled sgRNA scaffold. |
| Native Gel System | For EMSA to visually confirm complex formation and check for aggregation. | 6-8% Polyacrylamide gels with Mg2+-containing buffers. |
| Electroporation/Nucleofection Kit | For efficient delivery of pre-assembled RNP into hard-to-transfect primary cells. | Cell-type specific kits (e.g., Lonza P3, Neon System). |
| ITC or SPR Instrumentation | For label-free, rigorous thermodynamic analysis of binding interactions (kon, koff, ΔH). | MicroCal ITC, Biacore SPR systems. |
| Cryo-EM Grids & Reagents | For high-resolution structural determination of Cas9-sgRNA and target DNA complexes. | UltraAuFoil grids, vitrobot, cryo-EM grade buffers. |
This application note addresses a central challenge in CRISPR-Cas9 therapeutic development: the disparity in ribonucleoprotein (RNP) stability and delivery efficiency between immortalized cell lines and primary human cells. Successful gene editing in primary cells (e.g., T-cells, hematopoietic stem cells, hepatocytes) is critical for ex vivo and in vivo therapies but is hampered by intrinsic cellular factors absent in standard lines like HEK293 or HeLa. This document, framed within the broader thesis on optimizing Cas9-sgRNA complex formation for primary cell editing, details the key variables and provides validated protocols to overcome these barriers.
Intrinsic Factors: A Comparative Analysis
The following intrinsic factors quantitatively impact RNP stability, cellular uptake, and ultimate editing efficiency.
Table 1: Comparative Analysis of Intrinsic Cellular Factors
| Intrinsic Factor | Typical Cell Lines (HEK293, HeLa) | Primary Cells (e.g., T-cells, HSCs) | Impact on RNP Stability/Delivery |
|---|---|---|---|
| Cell Membrane Composition | Simplified, often higher passive permeability. Cholesterol-to-phospholipid ratio ~0.3-0.4. | Complex, varied. Higher cholesterol content (ratio ~0.5-0.7 in resting T-cells). | Reduced passive diffusion and electroporation-mediated delivery in primary cells due to membrane rigidity. |
| Endosomal Activity & Trafficking | Often altered; may have impaired endosomal maturation. | Robust and highly regulated endo-lysosomal pathway. | Increased RNP degradation in primary cells; rapid lysosomal degradation post-internalization. |
| Innate Immune Sensing (e.g., cGAS-STING, TLRs) | Frequently attenuated (e.g., HEK293 STING-deficient). | Fully functional. Cytosolic DNA/RNA sensors active. | Can trigger interferon response in primary cells, altering cell state, reducing viability, and potentially degrading RNP components. |
| Cytosolic Nuclease Activity | Variable, often lower. | High, particularly in immune cells (e.g., TREX1, RNases). | Increased degradation of sgRNA and DNA repair templates in the cytosol of primary cells. |
| Metabolic State & Redox Environment | High glycolytic flux, often adapted to culture. | Quiescent (e.g., HSCs) or activated states; more oxidizing cytosol. | Affects stability of protein complexes; disulfide bond formation in Cas9 may be altered. |
| Cell Cycle Status | Largely asynchronous, rapidly cycling. | Often predominantly in G0/G1 (e.g., resting T-cells, HSCs). | Non-homologous end joining (NHEJ) is active throughout cycle, but homology-directed repair (HDR) is restricted to S/G2, impacting editing outcomes. |
Table 2: Quantitative Data on RNP Delivery Efficiency (Representative Values)
| Delivery Method | Cell Type | Reported Delivery Efficiency* (%) | Relative Editing Efficiency (NHEJ%) | Key Limiting Factor in Primary Cells |
|---|---|---|---|---|
| Electroporation (Neon/4D-Nucleofector) | HEK293 | 95-99 (GFP mRNA) | 70-90 | N/A |
| Primary Human T-cells | 80-90 (GFP mRNA) | 30-60 | Cytotoxicity, post-delivery nuclease activity | |
| Human CD34+ HSCs | 70-85 (GFP mRNA) | 20-50 | Sensitivity to osmotic/oxidative stress | |
| Lipid Nanoparticles (LNPs) | HeLa | 80-95 | 40-70 | N/A |
| Primary Hepatocytes | 50-75 | 15-40 | Endosomal entrapment & degradation | |
| Chemical Transduction (Cell-penetrating peptides) | Jurkat (Line) | 60-80 | 20-50 | N/A |
| Primary NK Cells | 20-40 | 5-20 | Low endosomal escape efficiency |
*Delivery efficiency often measured by co-transfected fluorescent reporter (GFP mRNA or protein).
Detailed Experimental Protocols
Protocol 1: Assessing RNP Stability in Cytosolic Extracts
Objective: Quantify the half-life of pre-formed Cas9-sgRNA RNP in cytosolic environments from cell lines vs. primary cells. Materials: Purified Cas9 protein, synthetic sgRNA, primary cells (e.g., PBMCs), matched cell line, cell lysis buffer (without detergents), heparin (nuclease inhibitor control). Procedure:
- Prepare Cytosolic Extracts: Wash 10^7 cells with cold PBS. Resuspend in hypotonic lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, protease inhibitors). Incubate on ice 15 min, homogenize with 20 strokes in a Dounce homogenizer. Centrifuge at 10,000×g for 15 min at 4°C. Collect supernatant (cytosolic extract). Determine protein concentration.
- Form Fluorescently-labeled RNP: Label Cas9 with a fluorescent dye (e.g., Alexa Fluor 647) using a commercial labeling kit. Complex with sgRNA at a 1:1.2 molar ratio in PBS for 10 min at 25°C.
- Stability Assay: Mix 200 nM labeled RNP with 2 μg/μL of cytosolic extract in reaction buffer. Incubate at 37°C. Remove aliquots at 0, 5, 15, 30, 60, 120 min.
- Analysis: Run aliquots on native PAGE gel. Visualize fluorescence signal (Cy5 channel). Quantify intact RNP band intensity over time to calculate decay rate.
Protocol 2: Electroporation-Based RNP Delivery to Primary T-cells
Objective: Achieve high-efficiency gene editing in primary human T-cells with minimal cytotoxicity. Materials: Human primary T-cells (isolated via negative selection), P3 Primary Cell 4D-Nucleofector X Kit (Lonza), Cas9 protein (Alt-R S.p.), synthetic sgRNA (Alt-R), pre-warmed RPMI-1640+10% FBS. Procedure:
- RNP Formation: Complex 60 pmol of Cas9 protein with 72 pmol of sgRNA (1:1.2 ratio) in Duplex Buffer. Incubate at 25°C for 10 min.
- Cell Preparation: Islate and count T-cells. Centrifuge 1-2×10^6 cells, resuspend in 100 μL of room temperature Nucleofector Solution P3.
- Electroporation: Add 10 μL of formed RNP to cell suspension. Transfer to a certified cuvette. Electroporate using the 4D-Nucleofector with program EH-115. Immediately add 500 μL of pre-warmed medium.
- Post-Transfection Recovery: Transfer cells to a 24-well plate pre-filled with 500 μL warm medium. Incubate at 37°C, 5% CO2. Assess viability at 24h via trypan blue. Harvest cells at 48-72h for genomic DNA extraction and editing analysis via T7E1 assay or NGS.
Protocol 3: Measuring Endosomal Escape Efficiency
Objective: Quantify the fraction of internalized RNP that reaches the cytosol. Materials: pH-sensitive dye (e.g., pHrodo conjugated to streptavidin), biotinylated Cas9, sgRNA, primary cells, confocal microscopy/flow cytometry. Procedure:
- Prepare pH-Sensitive RNP: Biotinylate Cas9. Form RNP with sgRNA. Label RNP complex with pHrodo-streptavidin (pHrodo fluorescence increases in acidic endosomes).
- Internalization: Incubate cells with labeled RNP (100 nM) for 1-4h at 37°C or 4°C (negative control).
- Quench External Signal: Wash cells with cold PBS containing trypan blue (0.2%) to quench extracellular fluorescence.
- Image & Analyze: Using confocal microscopy, count total fluorescent puncta (endosomal) vs. diffuse cytosolic signal. Alternatively, use flow cytometry to measure total pHrodo signal. Treat parallel samples with endosomolytic agent (e.g., chloroquine) as a control for maximal escape; calculate efficiency as a percentage of this maximum.
Diagrams and Workflows
Diagram Title: RNP Delivery Pathways in Primary vs. Cultured Cells
Diagram Title: Primary Cell RNP Electroporation Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Primary Cell RNP Editing Research
| Reagent/Material | Supplier Examples | Function & Rationale |
|---|---|---|
| Alt-R S.p. Cas9 Nuclease V3 | Integrated DNA Technologies (IDT) | High-activity, recombinant Cas9 protein; low endotoxin, optimized for RNP formation with synthetic sgRNA. |
| Alt-R CRISPR-Cas9 sgRNA | IDT | Chemically modified synthetic sgRNA (2'-O-methyl, phosphorothioate) to enhance nuclease resistance and stability in primary cell cytosol. |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | Optimized buffer/electroporation cuvette system for sensitive primary cells (T-cells, HSCs). Maximizes viability and delivery. |
| CellTrace Violet / CFSE | Thermo Fisher Scientific | Cell proliferation dyes to track cell divisions post-editing, crucial as editing outcomes can be cell-cycle dependent. |
| Recombinant Human IL-2, IL-7, IL-15 | PeproTech | Cytokines for primary T-cell/NK cell activation and culture maintenance post-electroporation to ensure recovery. |
| Endosomolytic Agent (Chloroquine) | Sigma-Aldrich | Used experimentally to enhance endosomal escape; a control for assessing maximum possible delivery efficiency. |
| TREX1 Inhibitor (e.g., G150) | MedChemExpress | Small molecule inhibitor of cytosolic nuclease TREX1; can be used to test if nuclease activity limits editing efficiency. |
| Annexin V Apoptosis Detection Kit | BioLegend | To quantify apoptosis post-RNP delivery, distinguishing between general toxicity and editing-related cell death. |
| STING Inhibitor (e.g., H-151) | Cayman Chemical | To suppress innate immune sensing via the cGAS-STING pathway, improving viability in sensitive primary cells. |
Within the broader thesis on optimizing Cas9-sgRNA complex formation for primary cell editing, the precise design of the single-guide RNA (sgRNA) emerges as the most critical determinant of success. Primary cells, with their limited expansion capacity and sensitivity to off-target effects and cytotoxicity, demand sgRNAs that ensure high-fidelity complex formation. This application note details the essential sgRNA features and provides validated protocols for designing and testing sgRNAs to achieve efficient and specific editing in challenging primary cell models.
Essential sgRNA Features for High-Fidelity Complexes
High-fidelity complex formation requires an sgRNA that promotes stable Cas9 binding and precise DNA targeting while minimizing off-target interactions. The key features are quantitatively summarized below.
Table 1: Quantitative Parameters for Optimal sgRNA Design
| Feature | Optimal Parameter/Rule | Impact on Fidelity & Efficiency | Rationale for Primary Cells |
|---|---|---|---|
| Seed Region (PAM-proximal) | Nucleotides 1-12, high specificity. | Very High | Dictates initial DNA recognition; mismatches here severely reduce cleavage but can promote off-target binding if suboptimal. |
| GC Content | 40-60% | High | Affects sgRNA stability and secondary structure; impacts complex formation kinetics. Extremes reduce efficiency. |
| sgRNA Length | 20 nt spacer (standard) | Medium | Standard length balances specificity and activity. Truncated sgRNAs (17-18 nt, "tru-gRNAs") can enhance specificity. |
| Off-Target Prediction Score | CFD (Cutting Frequency Determination) score < 0.05; MIT Specificity Score > 90. | Critical | Predicts potential off-target sites. Low CFD/high MIT scores correlate with higher specificity, crucial for primary cell genomic integrity. |
| 5' Terminal Nucleotide | Guanosine (G) or Adenosine (A) preferred for U6 promoters. | High | U6 RNA Polymerase III requires a purine at the transcription start for high expression. |
| Secondary Structure | Minimal free energy (MFE) > -5 kcal/mol for spacer. | Medium | Internal structure in the spacer region can impede Cas9 binding, reducing on-target efficiency. |
Protocol 1:In SilicoDesign and Selection of High-Fidelity sgRNAs
Objective: To computationally design and rank sgRNAs targeting a gene of interest for high on-target efficiency and minimal off-target risk. Materials: "Research Reagent Solutions" table below. Workflow:
- Input: Provide the genomic DNA sequence of the target locus (500-1000 bp surrounding the target site) in FASTA format.
- Identify Candidates: Use the Cas9 Designer tool (e.g., from Tool A) to scan both DNA strands for all NGG PAM sites. Generate all possible 20-nt spacer sequences preceding each PAM.
- Filter by Sequence Rules: Eliminate sgRNAs with a non-purine 5' terminus or GC content outside 40-60%.
- Predict On-Target Efficiency: Score remaining sgRNAs using the Algorithm B (e.g., Doench '16/Rule Set 2). Select the top 5 candidates with scores > 60.
- Predict Off-Target Sites: For each top candidate, perform a genome-wide search allowing up to 3-4 mismatches using the Tool C algorithm. Calculate the CFD score for each potential off-target.
- Final Selection: Prioritize the sgRNA with the highest on-target score and the lowest cumulative CFD score for off-targets, particularly those with mismatches in the seed region.
The Scientist's Toolkit: Research Reagent Solutions for sgRNA Design & Testing
| Item | Function & Rationale |
|---|---|
| U6-Promoter driven sgRNA Cloning Vector | Backbone for expressing sgRNA in mammalian cells; contains selection marker (e.g., puromycin) for stable cell line generation. |
| High-Fidelity Cas9 Expression Plasmid | Source of SpCas9 protein. Use fidelity-enhanced variants (e.g., SpCas9-HF1, eSpCas9(1.1)) for primary cell work to reduce off-targets. |
| Primary Cell Nucleofection Kit | Specialized reagents for efficient, low-toxicity delivery of RNP or plasmids into sensitive primary cells. |
| T7 Endonuclease I (T7E1) or Surveyor Nuclease | Enzymes for detecting indel mutations via mismatch cleavage of heteroduplex PCR products. |
| NGS-based Off-Target Analysis Kit | Comprehensive kit for targeted deep sequencing of predicted and genome-wide off-target sites (e.g., GUIDE-seq, CIRCLE-seq). |
Protocol 2: Experimental Validation of sgRNA Fidelity in Primary Cells
Objective: To experimentally assess the on-target editing efficiency and specificity of a selected sgRNA in primary human T cells. Methodology: Ribonucleoprotein (RNP) nucleofection. Procedure:
- sgRNA Preparation: Synthesize the selected sgRNA sequence as a chemically modified, crRNA:tracrRNA duplex or as a single transcript. Resuspend in nuclease-free duplex buffer.
- RNP Complex Formation: For one reaction, combine 5 µg (≈ 30 pmol) of high-fidelity Cas9 protein with 7.5 µg (≈ 45 pmol) of sgRNA. Incubate at room temperature for 10-20 minutes to form the active RNP complex.
- Primary Cell Preparation: Isolate and activate primary human CD4+ T cells from buffy coats using CD3/CD28 beads. Culture for 48-72 hours.
- Nucleofection: Wash 1x10^6 cells. Resuspend cells in 100 µL of primary cell nucleofection solution. Mix with the pre-formed RNP complex. Transfer to a nucleofection cuvette and electroporate using the recommended program (e.g., Lonza 4D-Nucleofector, program EH-115).
- Post-Transfection Recovery: Immediately add pre-warmed culture medium and transfer cells to a plate. Analyze cells at 72-96 hours post-nucleofection.
- On-Target Analysis: Extract genomic DNA. PCR-amplify the target region (~500 bp). Quantify indel efficiency via T7E1 assay or, preferably, by NGS amplicon sequencing.
- Off-Target Validation: Perform targeted PCR amplification of the top 5-10 predicted off-target loci from Protocol 1, Step 5. Analyze by NGS to quantify indel frequencies (should be <0.5% for a high-fidelity design).
Diagrams
Title: Computational sgRNA Design and Selection Workflow
Title: RNP Complex Formation and Delivery to Primary Cells
This application note is framed within the broader thesis of optimizing Cas9 protein-sgRNA ribonucleoprotein (RNP) complex formation for genome editing in primary cells. The choice of Cas9 variant is a critical determinant of editing efficiency, specificity, and cellular viability in these sensitive, non-immortalized systems. This document compares Wild-Type (SpCas9), High-Fidelity (SpCas9-HF1, eSpCas9), and Nickase (SpCas9n) variants, providing quantitative data and detailed protocols for their application.
Comparative Analysis of Cas9 Variants
Table 1: Key Characteristics of Cas9 Variants for Primary Cell Editing
| Feature | Wild-Type SpCas9 | High-Fidelity (e.g., SpCas9-HF1) | Nickase (SpCas9n) |
|---|---|---|---|
| DNA Cleavage Mechanism | Blunt DSB | Blunt DSB | Single-strand break (nick) |
| Typical On-Target Efficiency | High (60-80% in amenable lines) | Moderately Reduced (50-70% of WT) | Very Low as single agent; requires pair for DSB |
| Off-Target Rate | High (frequent sgRNA-dependent) | Significantly Reduced (≥85% reduction) | Extremely Low (single nick is repaired faithfully) |
| Primary Cell Viability | Moderate (p53 response, apoptosis risk) | Improved (reduced toxic off-targeting) | High (minimal genotoxicity) |
| Optimal Use Case | Robust knockout where off-targets are less concerning | Therapeutic knockouts or sensitive functional genomics | Precise edits with paired nickase or base editor fusions |
| Common Delivery Format | RNP (pre-complexed) | RNP | RNP |
Table 2: Quantitative Performance Summary from Recent Studies (2023-2024)
| Parameter | Wild-Type SpCas9 RNP | SpCas9-HF1 RNP | Paired SpCas9n RNP (for DSB) |
|---|---|---|---|
| Indel Efficiency in T Cells | 75% ± 12% | 58% ± 15% | 40% ± 10% (spacing-dependent) |
| Cell Viability (Day 3 post-editing) | 65% ± 8% | 82% ± 7% | 90% ± 5% |
| Relative Off-Target Indels (by GUIDE-seq) | 1.0 (Reference) | 0.05 - 0.15 | Not Detectable (single nick) |
| HDR Efficiency (with donor) | 20-30% | 15-25% | 10-20% (paired nicking) |
Detailed Experimental Protocols
Protocol 3.1: RNP Complex Assembly for Primary Cell Electroporation
Application: Formation of active Cas9-sgRNA complexes for all variants. Materials: See "Scientist's Toolkit" (Section 5). Procedure:
- sgRNA Preparation: Resuscribe chemically modified sgRNA in nuclease-free duplex buffer (IDT) to 160 µM.
- RNP Complexing: In a sterile LoBind tube, combine:
- Cas9 protein (variant of choice): Final amount 60 pmol.
- sgRNA (target-specific): Final amount 80 pmol (1.33:1 sgRNA:Cas9 ratio).
- Nuclease-Free Buffer: To a total volume of 10 µL.
- Mix gently by pipetting. Do not vortex.
- Incubate at room temperature for 10-20 minutes to allow complete RNP formation.
- Primary Cell Preparation: During incubation, harvest and wash primary cells (e.g., T cells, HSCs) in appropriate electroporation buffer (e.g., P3 buffer for 4D-Nucleofector).
- Electroporation: Mix 10 µL RNP complex with 2-5 µL of 100 µM HDR template (if applicable) and 1e5-1e6 cells in 20 µL total electroporation volume. Transfer to cuvette/strip. Electroporate using a primary cell-optimized program (e.g., EO-115 for T cells).
- Recovery: Immediately add pre-warmed culture medium and transfer cells to a pre-coated culture plate. Assay at 48-72 hours.
Protocol 3.2: Off-Target Assessment by GUIDE-seq in Primary Cells
Application: Empirical determination of off-target sites for Wild-Type vs. Hi-Fi Cas9. Procedure:
- GUIDE-seq Oligonucleotide Delivery: Co-electroporate RNP complexes (Protocol 3.1) with 100 pmol of phosphorothioate-modified GUIDE-seq oligo (dsODN) per 1e5 cells.
- Genomic DNA Extraction: Harvest cells at day 3. Extract gDNA using a silica-column kit.
- Library Preparation: Fragment 1.5 µg gDNA by sonication (Covaris). Prepare sequencing libraries using a standard kit (e.g., NEBNext Ultra II), incorporating PCR steps with GUIDE-seq-specific primers to enrich for integration events.
- Sequencing & Analysis: Perform paired-end 150bp sequencing on an Illumina platform. Analyze reads using the open-source GUIDE-seq analysis software to map dsODN integration sites and identify off-target cleavages.
Diagrams
Title: Cas9 Variant Selection Workflow for Primary Cells
Title: Cas9-sgRNA Complex Formation and Variant Action
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Cas9 RNP Editing in Primary Cells
| Item | Function & Key Consideration |
|---|---|
| Recombinant Cas9 Protein (WT, Hi-Fi, Nickase) | The core nuclease. Hi-Fi variants (SpCas9-HF1, eSpCas9) reduce off-targets. Alt-R S.p. Cas9 Nuclease V3 is common. |
| Chemically Modified sgRNA (synthetictracrRNA + crRNA) | Enhances stability and reduces immune activation in primary cells. Use Alt-R CRISPR-Cas9 sgRNA or similar. |
| Electroporation System (4D-Nucleofector, Lonza) | Gold-standard for primary cell RNP delivery. Use cell-type-specific programs and buffers (e.g., P3 Kit). |
| Cell Culture Media (X-VIVO 15, TexMACS) | Serum-free, cytokine-supplemented media optimized for primary immune cells post-electroporation. |
| HDR Template (ssODN or dsDNA) | For precise edits. Use high-purity, HPLC-purified single-stranded oligos with homology arms (90-120 nt). |
| Viability Dye (e.g., 7-AAD) | For accurate flow cytometry assessment of post-editing survival. |
| Genomic DNA Extraction Kit (Silica-column based) | For clean gDNA prep post-editing for sequencing assays (T7E1, NGS). |
| NGS Library Prep Kit (e.g., NEBNext Ultra II) | For deep sequencing of target loci to quantify editing efficiency and specificity. |
Step-by-Step Protocols: Assembling and Delivering Cas9-sgRNA Complexes into Primary Cells
Within the context of a broader thesis on Cas9 protein sgRNA complex formation for primary cell editing research, the choice of CRISPR-Cas9 delivery modality is paramount. Pre-assembled Cas9-sgRNA ribonucleoprotein (RNP) complexes and plasmid-based expression systems represent two fundamentally different strategies, each with distinct implications for editing efficiency, specificity, cellular toxicity, and translational potential. This guide provides a detailed comparison through application notes and protocols for researchers, scientists, and drug development professionals.
Quantitative Comparison
Table 1: Core Characteristics and Performance Metrics
| Parameter | Pre-assembled RNP Delivery | Plasmid/DNA Delivery |
|---|---|---|
| Time to Onset of Activity | Minutes to hours (immediate activity) | 12-48 hours (requires transcription/translation) |
| Duration of Activity | Short (24-72 hours, degrades rapidly) | Prolonged (days to weeks, persistent expression) |
| Editing Efficiency (Typical in Primary Cells) | 40-80% | 10-40% |
| Off-target Effect Incidence | Lower (limited exposure window) | Higher (sustained Cas9/gRNA presence) |
| Cellular Toxicity | Lower (minimizes immune activation, no DNA integration) | Higher (TLR9/immune activation, risk of genomic integration) |
| Ease of Multiplexing | Moderate (requires co-delivery of multiple RNPs) | High (multiple gRNAs encoded on single plasmid) |
| Manufacturing Complexity | Higher (protein purification, complex assembly) | Lower (standard molecular biology) |
| Cost per Experiment | Higher | Lower |
| Regulatory/Translational Path | More favorable (defined composition, no foreign DNA) | Complicated (persisting genetic material) |
Table 2: Application-Specific Suitability
| Research Context | Recommended Method | Rationale |
|---|---|---|
| Primary Cell Editing (T cells, HSCs, iPSCs) | Pre-assembled RNP | High efficiency, low toxicity, minimal culture time. |
| In vivo Gene Therapy | Pre-assembled RNP (via viral/non-viral delivery) | Reduced immunogenicity, controlled exposure. |
| High-Throughput Screening | Plasmid/Lentiviral | Stable integration, scalable library delivery. |
| Disease Modeling (Requiring Stable Line) | Plasmid/Viral | Selection and clonal expansion possible. |
| Rapid Functional Knockout Assay | Pre-assembled RNP | Fast, transient, high efficiency. |
Detailed Protocols
Protocol 1: Production and Delivery of Pre-assembled Cas9 RNP for Primary T Cell Editing
Objective: To achieve high-efficiency, low-toxicity gene knockout in primary human T cells via electroporation of Cas9 RNP complexes.
Key Research Reagent Solutions:
- Recombinant Cas9 Nuclease: High-purity, endotoxin-free S. pyogenes Cas9 protein.
- Synthetic sgRNA: Chemically modified, HPLC-purified sgRNA targeting gene of interest.
- Electroporation Buffer/System: Optimized for primary immune cells (e.g., P3 buffer with 4D-Nucleofector).
- Primary Human T Cells: Isolated from peripheral blood mononuclear cells (PBMCs), activated for 2-3 days.
- RNase Inhibitor: To protect RNP integrity during assembly.
- Flow Cytometry Antibodies: For assessing editing efficiency via knockout protein loss.
Procedure:
- RNP Complex Assembly: In a nuclease-free tube, combine 60 pmol of Cas9 protein with 120 pmol of synthetic sgRNA (2:1 molar ratio of sgRNA:Cas9) in a total volume of 5 µL of sterile duplex buffer (e.g., 30 mM HEPES, 100 mM KCl). Mix gently and incubate at room temperature for 10-20 minutes.
- T Cell Preparation: Harvest activated T cells, wash with PBS, and count. Resuspend cells at a concentration of 1-2 x 10^7 cells per 100 µL of pre-warmed electroporation buffer.
- Electroporation: Add 5 µL of assembled RNP to 100 µL of cell suspension in an electroporation cuvette. Mix gently. Electroporate using a pre-optimized program for primary T cells (e.g., "EO-115" on a 4D-Nucleofector X Unit). Immediately add 500 µL of pre-warmed culture medium supplemented with cytokines (e.g., IL-2) to the cuvette.
- Post-Electroporation Culture: Transfer cells to a pre-warmed culture plate. Analyze editing efficiency at 48-72 hours post-electroporation via flow cytometry (for protein knockout) or next-generation sequencing (for indel analysis).
Protocol 2: Plasmid-Based Cas9 Delivery to Primary Cells via Nucleofection
Objective: To deliver Cas9 and sgRNA via plasmid DNA to primary cells for applications requiring sustained expression or stable integration.
Key Research Reagent Solutions:
- Cas9/sgRNA Expression Plasmid: Plasmid encoding Cas9 and sgRNA(s) under mammalian promoters (e.g., U6 for sgRNA, EF1α for Cas9).
- Endotoxin-Free Plasmid Prep Kit: For high-purity plasmid isolation.
- Electroporation Kit for Primary Cells: Optimized for DNA delivery.
- Selection Antibiotics/Puromycin: If plasmid contains a selectable marker for enrichment.
- qPCR Reagents: For quantifying plasmid copy number or Cas9 expression levels.
Procedure:
- Plasmid Preparation: Purify the Cas9/sgRNA plasmid using an endotoxin-free maxiprep kit. Resuspend DNA in nuclease-free TE buffer or water. Quantify by spectrophotometry (260/280 ratio >1.8).
- Cell Preparation: As per Protocol 1, harvest and wash primary cells (e.g., T cells, HSCs). Resuspend at 1-2 x 10^7 cells per 100 µL of appropriate DNA-specific electroporation buffer.
- Electroporation: Add 2-5 µg of plasmid DNA to the cell suspension. Electroporate using a program optimized for plasmid DNA delivery to the specific primary cell type (often different from RNP programs).
- Recovery and Analysis: Recover cells as in Step 4 of Protocol 1. Due to delayed expression, assess editing efficiency no earlier than 72 hours post-electroporation, typically at 96-120 hours. Monitor cells for prolonged periods for phenotypic assays.
Visualizations
Workflow of CRISPR Delivery Methods in Primary Cells
Timeline Comparison of RNP vs Plasmid Protocols
The Scientist's Toolkit
Table 3: Essential Research Reagents for Cas9 Complex Formation & Delivery
| Reagent/Material | Function | Example Product/Note |
|---|---|---|
| Recombinant Cas9 Protein | The CRISPR effector nuclease. Must be high purity and endotoxin-free for sensitive primary cells. | Alt-R S.p. Cas9 Nuclease V3, TruCut Cas9 Protein. |
| Synthetic sgRNA | Chemically modified single-guide RNA for target specificity and enhanced stability. | Alt-R CRISPR-Cas9 sgRNA, Synthego sgRNA. |
| Cas9 Expression Plasmid | Mammalian vector for constitutive or inducible expression of Cas9 and sgRNA(s). | px458 (Addgene #48138), lentiCRISPRv2. |
| Electroporation/Nucleofector System | Enables physical delivery of macromolecules (RNPs, plasmids) into primary cells. | Lonza 4D-Nucleofector X Unit, Neon Transfection System (Thermo). |
| Cell-Specific Electroporation Kits | Optimized buffers and cuvettes for specific cell types (T cells, HSCs, iPSCs). | P3 Primary Cell Kit, Human T Cell Kit. |
| RNase Inhibitor | Protects sgRNA and RNP complexes from degradation during assembly. | Recombinant RNase Inhibitor. |
| NGS-based Editing Analysis Service | Quantifies on-target indel efficiency and off-target profiling. | Illumina MiSeq amplicon sequencing, IDT xGen NGS services. |
| Flow Cytometry Antibodies | For assessing protein-level knockout efficiency in edited cell populations. | Antibodies against target cell surface or intracellular protein. |
| Cytokines/Growth Factors | Maintains primary cell viability and proliferation during post-editing recovery. | Recombinant human IL-2 (for T cells), SCF/Flt3L/TPO (for HSCs). |
Thesis Context: This work is part of a broader thesis investigating the biophysical parameters governing Cas9 protein:sgRNA ribonucleoprotein (RNP) complex formation and stability to achieve maximal on-target editing efficiency and minimize off-target effects in hard-to-transfect primary cells, a critical step for therapeutic development.
Achieving high-efficiency genome editing in primary cells (e.g., T cells, hematopoietic stem cells, neurons) is paramount for research and drug development. Electroporation/nucleofection of pre-assembled Cas9:sgRNA RNP complexes is the gold standard due to its rapid activity and reduced off-target risk. The molar ratio of Cas9 to sgRNA during complex assembly is a critical, often optimized variable influencing RNP stability, cellular delivery, and ultimate editing outcomes.
Table 1: Reported Optimal Cas9:sgRNA Molar Ratios for Primary Cell Types
| Primary Cell Type | Nucleofection System/Kit | Reported Optimal Cas9:sgRNA Molar Ratio | Key Editing Outcome (Metric) | Citation (Year) |
|---|---|---|---|---|
| Human T Cells | Lonza 4D-Nucleofector, P3 Kit | 1:2 to 1:3 | >70% INDELs at TRAC locus | Roth et al. (2018) |
| Human CD34+ HSPCs | Lonza 4D-Nucleofector, P3 Kit | 1:2.5 | ~60% INDELs at HBB locus | DeWitt et al. (2016) |
| Human iPSC-derived Neurons | Bio-Rad Gene Pulser Xcell | 1:1.5 | ~45% Knock-in efficiency | Lin et al. (2022) |
| Mouse Bone Marrow Dendritic Cells | Neon Transfection System | 1:2 | ~55% protein knockout (flow) | Suresh et al. (2023) |
| Primary Human Keratinocytes | Amaxa Nucleofector | 1:3 | ~40% GFP reporter integration | Byrne et al. (2024) |
Table 2: Impact of Ratio Deviation from Optimum
| Cas9:sgRNA Ratio | Observed Effect on RNP Complex | Typical Editing Outcome vs. Optimal |
|---|---|---|
| Sub-optimal (e.g., 1:0.5) | sgRNA-limiting, excess free Cas9 | Significantly reduced editing. Free Cas9 may compete for delivery, increase cellular toxicity. |
| Optimal (e.g., 1:2 - 1:3) | Fully assembled, sgRNA-stabilized RNP with minimal free components. | Maximal on-target editing. Efficient nuclear entry and target DNA saturation. |
| Supra-optimal (e.g., 1:5+) | sgRNA-saturating, excess free sgRNA. | Potential for reduced editing or increased variability. Excess sgRNA may interfere with RNP cellular uptake or promote off-target binding. |
Core Experimental Protocol: RNP Assembly & Nucleofection for T Cells
A. Materials & Reagent Preparation
- Cas9 Nuclease: High-purity, recombinant S. pyogenes Cas9 protein (e.g., IDT Alt-R S.p. Cas9 Nuclease V3).
- sgRNA: Chemically synthesized, HPLC-purified synthetic sgRNA with modified ends (e.g., Alt-R CRISPR-CrRNA and tracrRNA) or in vitro transcribed (IVT), purified sgRNA.
- Nucleofection Buffer: Cell-type specific kit (e.g., Lonza P3 Primary Cell 4D-Nucleofector X Kit).
- Electroporation Cuvettes/Strips: As required by the system.
- Cell Culture Media: Pre-warmed, complete growth media supplemented with appropriate cytokines (e.g., IL-2 for T cells).
B. Step-by-Step Protocol
- Cell Preparation: Isolate and activate primary human T cells. Wash and resuspend in appropriate media without antibiotics. Count and aliquot 1x10^6 cells per nucleofection condition.
- RNP Complex Assembly:
- Dilute Cas9 protein and sgRNA separately in nuclease-free duplex buffer (e.g., IDT) or PBS.
- For a 1:2.5 molar ratio: Combine 5 µg (approx. 31.5 pmol) of Cas9 protein with a molar excess of sgRNA (78.75 pmol). Total reaction volume should be ≤10 µL.
- Mix gently by pipetting. Incubate at room temperature for 10-20 minutes to allow complete RNP formation.
- Nucleofection Setup:
- Transfer the assembled RNP complex (10 µL) to a nucleofection cuvette/strip well.
- Pellet the 1x10^6 cells, completely aspirate the supernatant.
- Resuspend the cell pellet in 100 µL of pre-aliquoted nucleofection solution from the kit.
- Immediately transfer the cell suspension to the cuvette/strip well containing the RNP. Mix gently by pipetting 2-3 times.
- Electroporation:
- Place the cuvette in the nucleofector and run the cell-type-specific pre-programmed code (e.g., "EO-115" for human T cells using the P3 kit).
- Post-Nucleofection Recovery:
- Immediately after the pulse, add 500 µL of pre-warmed, complete culture media to the cuvette.
- Gently transfer the cells to a culture plate containing pre-warmed media.
- Culture cells at 37°C, 5% CO₂. Assess viability and editing efficiency after 48-72 hours.
Diagrams
Title: RNP Assembly Ratio Determines Editing Outcome
Title: Primary Cell RNP Nucleofection Workflow
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for Primary Cell RNP Editing
| Item | Function & Importance | Example Product/Brand |
|---|---|---|
| Recombinant Cas9 Nuclease | The core editing enzyme. High purity and activity are critical for efficiency and low toxicity. | IDT Alt-R S.p. Cas9 Nuclease V3; Thermo Fisher TrueCut Cas9 Protein v2. |
| Chemically Modified sgRNA | Synthetic RNA with phosphorothioate and 2'-O-methyl modifications enhances nuclease resistance and RNP stability. | IDT Alt-R CRISPR-CrRNA & tracrRNA; Synthego SYNTHEGO sgRNA. |
| Cell-Type Specific Nucleofection Kit | Buffer solutions optimized for specific primary cell types to maximize viability and delivery efficiency. | Lonza 4D-Nucleofector X Kit (P3, SE, etc.); Thermo Fisher Neon Transfection System Kit. |
| Nuclease-Free Duplex Buffer | Optimized ionic solution for proper Cas9:sgRNA hybridization and stable RNP formation. | IDT Duplex Buffer; Teknova Nuclease-Free Buffer. |
| Cell Activation & Culture Reagents | Cytokines and media formulations essential for primary cell health, recovery, and proliferation post-nucleofection. | IL-2, IL-7, IL-15 for T cells; StemSpan for HSCs. |
| Genome Editing Detection Reagents | For quantifying editing efficiency (INDELs, HDR). Essential for protocol optimization. | IDT Alt-R Genome Editing Detection Kit (T7E1); NGS-based amplicon sequencing kits. |
This protocol details the in vitro reconstitution of functional Cas9:sgRNA ribonucleoproteins (RNPs) for genome editing applications. Within the broader thesis on "Mechanisms of Cas9-sgRNA Complex Formation for Enhanced Primary Cell Editing," this methodology is foundational. Efficient, pre-assembled RNPs offer advantages over plasmid-based delivery, including rapid kinetics, reduced off-target effects, and applicability to hard-to-transfect primary cells. This document provides a standardized, reproducible process for generating high-purity, active RNPs suitable for sensitive downstream research in drug development and cellular therapy.
Key Reagent Solutions and Materials
Table 1: Research Reagent Solutions for RNP Assembly & Purification
| Reagent/Material | Function in Protocol | Example Source/Notes |
|---|---|---|
| Recombinant Cas9 Nuclease | The effector protein; SpCas9 (or variants like HiFi Cas9) is commonly used. Must be nuclease-grade, endotoxin-free. | Purified in-house or commercial sources (IDT, Thermo Fisher). |
| Chemically Synthesized sgRNA | Guides Cas9 to specific genomic DNA sequence. Requires full chemical modification (2'-O-methyl, phosphorothioate) for stability in primary cells. | Synthesized via solid-phase (e.g., HPLC-purified from commercial vendors). |
| RNase Inhibitor | Protects sgRNA from degradation during assembly and purification steps. | Murine or human recombinant (e.g., RNasin Plus). |
| Assembly Buffer (1X) | Optimized ionic and pH conditions for proper RNP folding and stability. Typically contains HEPES, KCl, MgCl2, DTT, glycerol. | 20 mM HEPES-KOH (pH 7.5), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 10% glycerol. |
| Size-Exclusion Chromatography (SEC) Column | Purifies assembled RNP from free sgRNA, free protein, and aggregates. | HiLoad 16/600 Superdex 200 pg or comparable. |
| Fast Protein Liquid Chromatography (FPLC) System | For precise, automated purification via SEC. | ÄKTA pure or similar. |
| Amicon Ultra Centrifugal Filters | Concentrates purified RNP to working concentrations for electroporation or transfection. | 100 kDa molecular weight cut-off (MWCO). |
| Nuclease-Free Water/Buffers | Prevents RNA degradation in all steps. | Certified, DEPC-treated. |
Detailed Protocol for RNP Assembly and Purification
Pre-Assembly Preparation
- Thaw all components on ice: Cas9 protein, sgRNA (resuspended in nuclease-free water), 5X Assembly Buffer, RNase Inhibitor.
- Prepare 1X Assembly Buffer by diluting 5X stock with nuclease-free water. Filter through a 0.22 µm filter.
- Dilute sgRNA to a intermediate concentration (e.g., 100 µM) in nuclease-free water to ensure accurate pipetting.
In Vitro RNP Assembly
This protocol yields a 1:1.2 molar ratio complex, ensuring complete Cas9 saturation.
- In a sterile, nuclease-free microcentrifuge tube, combine the following on ice:
- Cas9 Protein: 100 pmol (e.g., 5 µL of 20 µM stock).
- sgRNA: 120 pmol (e.g., 4.8 µL of 25 µM stock).
- RNase Inhibitor: 1 µL (40 U/µL).
- 1X Assembly Buffer: to a final volume of 50 µL.
- Mix gently by pipetting up and down. Do not vortex.
- Incubate at 25°C (room temperature) for 10 minutes to allow complex formation.
- Transfer to ice. The assembled RNP is now ready for purification or can be used directly in some applications (with lower efficiency).
Purification by Size-Exclusion Chromatography (SEC)
Removes unbound sgRNA and protein, aggregates, and exchange into optimal buffer.
FPLC Method:
- Equilibrate the SEC column (e.g., Superdex 200 Increase 10/300 GL) with 1.5 column volumes (CV) of Degassed & Filtered SEC Buffer (e.g., 20 mM HEPES pH 7.5, 300 mM KCl, 1 mM MgCl2, 5% glycerol).
- Load the entire 50 µL assembled RNP mixture onto the column via a sample loop.
- Run Isocratic Elution at a flow rate of 0.5 mL/min, collecting 0.5 mL fractions.
- Monitor the UV absorbance at 260 nm (RNA/protein) and 280 nm (protein). The RNP complex will elute as a peak before free sgRNA (260 nm high) and free Cas9 (280 nm high).
Table 2: Expected SEC Elution Profile (Superdex 200 Increase)
| Peak | Approx. Elution Volume (mL) | A260/A280 Ratio | Identity |
|---|---|---|---|
| Void / Aggregates | 7.5 - 8.5 | Variable | Large aggregates (discard). |
| RNP Complex | 9.5 - 11.0 | ~1.2 | Functional Cas9:sgRNA complex. |
| Free sgRNA | 12.5 - 14.0 | >2.0 | Unbound guide RNA. |
| Free Cas9 | 13.5 - 15.0 | ~0.6 | Unbound protein. |
- Analyze Fractions by SDS-PAGE (for protein) and denaturing urea-PAGE (for RNA) to confirm co-elution.
- Pool the fractions containing the pure RNP complex (center of the peak).
Concentration and Quality Control
- Concentrate the pooled RNP using a 100 kDa MWCO centrifugal filter unit. Centrifuge at 4,000 x g at 4°C until desired volume (typically 20-50 µL) is reached.
- Determine Concentration: Measure A260 and A280.
- Use the calculated extinction coefficient for the specific RNP (ε(Cas9:sgRNA) ~ 1,000,000 M⁻¹cm⁻¹).
- Formula: RNP Concentration (M) = (A260) / (ε * path length (1 cm)).
- Aliquot the purified RNP, flash-freeze in liquid nitrogen, and store at -80°C. Avoid repeated freeze-thaw cycles.
- Functional Validation: Perform an in vitro cleavage assay using a PCR-amplified target DNA substrate. Incubate 50 nM RNP with 100 ng target DNA in 1X NEBuffer 3.1 at 37°C for 1 hour, then analyze by agarose gel electrophoresis for expected fragment sizes.
Visualizations
Diagram Title: RNP Assembly & Purification Workflow
Diagram Title: Protocol Role in Cas9 Primary Cell Research Thesis
The efficient delivery of CRISPR-Cas9 ribonucleoprotein (RNP) complexes into primary cells represents a critical bottleneck in therapeutic genome editing research. Unlike immortalized cell lines, primary cells are often fragile, non-dividing, and recalcitrant to standard transfection methods. This application note details three pivotal strategies—Electroporation, Nucleofection, and Novel Carrier systems—for achieving primary cell-specific RNP delivery, directly supporting thesis research on optimizing Cas9-sgRNA complex formation and editing efficiency in primary human T-cells and hematopoietic stem cells (HSCs).
Application Notes: Comparative Analysis of Delivery Strategies
Quantitative Comparison of Delivery Methods
The following table summarizes key performance metrics for each strategy, based on recent literature (2023-2024) focusing on primary human T-cells and CD34+ HSCs.
Table 1: Performance Metrics of RNP Delivery Strategies in Primary Cells
| Parameter | Electroporation (e.g., BTX ECM 830) | Nucleofection (4D-Nucleofector) | Lipid-Based Nanoparticles (Novel Carrier) | Polymer-Based Carriers (e.g., PGA-Oligoaminoamide) |
|---|---|---|---|---|
| Max Viability (T-cells) | 60-75% | 70-85% | 85-95% | 80-90% |
| Editing Efficiency | 40-60% | 50-80% | 30-50% | 20-45% |
| Throughput | Medium | High (96-well) | High | High |
| Cost per Sample | $$ | $$$ | $$ | $ |
| Specialized Equipment | Required | Required | Not Required | Not Required |
| Key Advantage | Broad applicability | High efficiency in hard-to-transfect | High viability, scalable | Tunable, potentially low immunogenicity |
| Key Limitation | High cytotoxicity | Cell-type specific optimization needed | Lower efficiency for RNP | Complexity in synthesis/formulation |
Research Reagent Solutions Toolkit
Table 2: Essential Materials for Primary Cell RNP Delivery Experiments
| Reagent/Material | Supplier Examples | Function in Protocol |
|---|---|---|
| Cas9 Nuclease, S. pyogenes (RNP ready) | IDT, Thermo Fisher, Synthego | The editing enzyme, pre-complexed with sgRNA to form the active RNP. |
| Chemically Modified sgRNA | Dharmacon, IDT | Enhances stability and reduces immunogenicity; critical for high-efficiency editing. |
| P3 Primary Cell 4D-Nucleofector Kit | Lonza | Cell-type specific nucleofection solution for HSCs and T-cells. |
| SF Cell Line 4D-Nucleofector Kit | Lonza | Optimized solution for certain sensitive primary cell types. |
| Opti-MEM Reduced Serum Medium | Thermo Fisher | Diluent for lipid nanoparticles; maintains cell health during transfection. |
| LipoJet (for RNP) Transfection Reagent | SignaGen Laboratories | Commercial lipid formulation specifically optimized for protein/RNP delivery. |
| Recombinant Human IL-2 | PeproTech | Critical for T-cell recovery and expansion post-electroporation/nucleofection. |
| StemSpan SFEM II | StemCell Technologies | Serum-free expansion medium for HSC culture post-editing. |
| Annexin V Apoptosis Detection Kit | BioLegend | To quantify cytotoxicity/viability post-delivery. |
| Genome Editing Detection Kit (T7E1) | NEB | To validate and quantify editing efficiency at the target locus. |
Detailed Experimental Protocols
Protocol: Nucleofection of Cas9 RNP into Primary Human T-Cells
Objective: Achieve high-efficiency gene knockout (e.g., PDCD1) in activated human CD3+ T-cells.
Materials:
- Primary human CD3+ T-cells, activated for 48-72h with CD3/CD28 beads.
- Cas9 protein (IDT, Alt-R S.p. HiFi Cas9 Nuclease V3).
- Alt-R CRISPR-Cas9 sgRNA (targeting PDCD1), chemically modified.
- Lonza P3 Primary Cell 4D-Nucleofector Kit (Solution, Supplement, Cuvettes).
- Lonza 4D-Nucleofector X Unit.
- Pre-warmed RPMI-1640 + 10% FBS + 100 U/mL IL-2.
Procedure:
- RNP Complex Formation: Combine 6 µg (60 pmol) of Cas9 protein with 3 µg (60 pmol) of sgRNA in a sterile microcentrifuge tube. Add nuclease-free duplex buffer to a final volume of 10 µL. Incubate at room temperature for 10 minutes.
- Cell Preparation: Harvest activated T-cells, count, and centrifuge. Resuspend cell pellet in pre-warmed Nucleofector Solution P3 to achieve a density of 1 x 10^7 cells per 100 µL.
- Nucleofection Mix: Combine 100 µL of cell suspension with the 10 µL pre-formed RNP complex. Transfer the entire 110 µL to a 100 µL Nucleofector cuvette, avoiding air bubbles.
- Nucleofection: Place cuvette in the 4D-Nucleofector X Unit and run the recommended program for primary human T-cells: EO-115.
- Recovery: Immediately after pulsing, add 500 µL of pre-warmed culture medium (RPMI+IL-2) to the cuvette. Gently transfer cells to a 12-well plate containing 1.5 mL of pre-warmed medium.
- Culture & Analysis: Incubate cells at 37°C, 5% CO2. Assess viability at 24h using trypan blue. Harvest cells at 72-96h post-nucleofection for genomic DNA extraction and analysis of editing efficiency via T7E1 assay or NGS.
Protocol: Electroporation of Cas9 RNP into CD34+ Hematopoietic Stem/Progenitor Cells (HSPCs)
Objective: Edit a therapeutic target (e.g., BCL11A enhancer) in human mobilized peripheral blood CD34+ cells.
Materials:
- Human CD34+ HSPCs (fresh or thawed).
- Cas9 protein (Thermo Fisher, TrueCut Cas9 Protein v2).
- Synthetic sgRNA (targeting the BCL11A enhancer).
- BTX ECM 830 Square Wave Electroporation System & 2mm gap cuvettes.
- Electroporation Buffer: Opti-MEM + 1% HSA.
- Serum-free StemSpan SFEM II medium with cytokines (SCF, TPO, FLT3L).
Procedure:
- RNP Complex Formation: Pre-complex 5 µg (50 pmol) Cas9 protein with 2.5 µg (50 pmol) sgRNA in a total volume of 20 µL nuclease-free buffer. Incubate 10 min at RT.
- Cell Preparation: Wash CD34+ cells twice in PBS and once in electroporation buffer. Resuspend at 2 x 10^7 cells/mL in electroporation buffer.
- Electroporation Mix: Mix 20 µL of cell suspension (400,000 cells) with 20 µL of pre-formed RNP. Transfer to a 2mm electroporation cuvette.
- Electroporation: Place cuvette in the holder and deliver a single square wave pulse: Voltage: 500V, Pulse Width: 2ms, Number of Pulses: 1.
- Immediate Recovery: Post-pulse, incubate cells in the cuvette for 10 minutes at room temperature. Gently resuspend and transfer to pre-warmed StemSpan SFEM II medium with cytokines.
- Culture & Analysis: Culture cells at 37°C, 5% CO2 at low density (<5x10^5 cells/mL). Assess viability at 24h. Perform genomic analysis at 48-72h and conduct functional assays (e.g., erythroid differentiation for BCL11A editing) at day 7-14.
Protocol: Lipid Nanoparticle (LNP) Formulation for RNP Delivery
Objective: Formulate ionizable lipid-based LNPs for low-cytotoxicity RNP delivery to primary hepatocytes.
Materials:
- Ionizable lipid (e.g., DLin-MC3-DMA), cholesterol, DSPC, DMG-PEG2000.
- Cas9 RNP complex (as prepared in 2.1).
- Microfluidic mixer (e.g., NanoAssemblr Ignite).
- ˙Acidified citrate buffer (pH 4.0).
- ˙1x PBS, pH 7.4.
- ˙100 kDa MWCO dialysis cassettes.
Procedure:
- Lipid Solution Preparation: Dissolve lipids in ethanol at molar ratios (Ionizable Lipid:Cholesterol:DSPC:DMG-PEG = 50:38.5:10:1.5) to a total lipid concentration of 12.5 mM.
- Aqueous Phase Preparation: Dilute the pre-formed Cas9 RNP complex in acidified citrate buffer (pH 4.0) to a final concentration of 100 µg/mL.
- Microfluidic Mixing: Load the lipid-ethanol solution and the RNP-acid buffer into separate syringes. Set the total flow rate (TFR) to 12 mL/min and a flow rate ratio (aqueous:organic) of 3:1. Initiate mixing.
- Buffer Exchange & Dialysis: Collect the formed LNP suspension and immediately dilute in 1x PBS (pH 7.4). Transfer to a dialysis cassette and dialyze against 2L of 1x PBS for 4 hours at 4°C, with one buffer change.
- Concentration & Characterization: Concentrate LNPs using Amicon Ultra centrifugal filters (100 kDa MWCO). Characterize particle size (Z-average ~80-100 nm) via DLS and measure RNP encapsulation efficiency using a Ribogreen assay.
- Cell Treatment: Treat primary human hepatocytes with LNP-RNPs at a final Cas9 concentration of 100-200 nM. Assess editing and viability at 72-96h.
Visualization of Workflows and Pathways
Diagram 1: Primary Cell RNP Delivery & Editing Workflow
Diagram 2: Mechanisms of Physical & Carrier-Based Delivery
Thesis Context: The efficient delivery and formation of the Cas9-sgRNA ribonucleoprotein (RNP) complex is the central bottleneck in achieving high-efficiency, low-toxicity genome editing in therapeutically relevant primary cells. This article details protocols and case studies that optimize this critical step across diverse, hard-to-transfect cell types.
Table 1: Editing Efficiency and Viability Across Primary Cell Types Using RNP Electroporation
| Cell Type | Target Gene | Delivery Method | Avg. Editing Efficiency (%) | Avg. Viability (%) | Key Application |
|---|---|---|---|---|---|
| Human T-Cells | TRAC | Neon Electroporation | 85-95 | 60-75 | CAR-T Cell Generation |
| Human CD34+ HSCs | BCL11A enhancer | 4D-Nucleofector (P3 Kit) | 70-80 | 40-60 | Sickle Cell Disease Therapy |
| Human iPSC-Derived Neurons | HTT | AAV-DJ with sgRNA | 40-60 (NHEJ) | >80 | Huntington's Disease Modeling |
| Primary Hepatocytes | PCSK9 | Lipid Nanoparticles (LNPs) | >90 in vivo | N/A | Hypercholesterolemia |
| Airway Stem Cells | CFTR | Adenoviral Vector (AVV) | ~30 (HDR) | ~70 | Cystic Fibrosis |
Table 2: RNP Complex Formation & Delivery Parameters
| Parameter | T-Cells | HSCs | Neurons | Rationale |
|---|---|---|---|---|
| Cas9:sgRNA Ratio | 1:2.5 | 1:3 | 1:2 (AAV co-delivery) | Minimizes free Cas9, optimizes complex saturation. |
| RNP Assembly Time | 10 min, 25°C | 15-20 min, 25°C | N/A (AAV) | Ensures complete complex formation prior to delivery. |
| Electroporation Buffer | P3 Primary Cell Solution | P3 Primary Cell Solution | N/A | Low ionic strength enhances RNP uptake during pulse. |
| Post-Electroporation Rest | 10 min, RT | Immediate culture | N/A | Allows membrane recovery before handling. |
Detailed Protocols
Protocol 1: High-EfficiencyTRACLocus Knock-in in Primary Human T-Cells
Objective: Generate universal CAR-T cells via RNP-mediated integration of a CAR cassette into the TRAC locus using an AAV6 donor template.
- RNP Complex Assembly:
- Resuspend 60 µg of purified SpCas9 protein and 12 µg (1:2.5 molar ratio) of chemically modified TRAC-targeting sgRNA (Synthego) in duplex buffer.
- Incubate at 25°C for 10 minutes.
- T-Cell Preparation:
- Isolate CD3+ T-cells from leukapheresis product using a Ficoll gradient and positive selection beads.
- Activate with CD3/CD28 antibodies for 48 hours in X-VIVO media with 100 IU/mL IL-2.
- Electroporation & HDR:
- Mix 2e6 activated T-cells with pre-formed RNP and 2e10 vg of AAV6 donor vector in 100 µL of P3 Primary Cell Solution (Lonza).
- Electroporate using the 4D-Nucleofector (X-Unit, program EH-115).
- Immediately transfer cells to pre-warmed, IL-2-supplemented media.
- Analysis:
- Assess viability at 24h (Trypan Blue).
- Quantify editing and knock-in efficiency at day 5 via flow cytometry (for surface CAR) and NGS of the target locus.
Protocol 2:BCL11AEnhancer Editing in Human CD34+ Hematopoietic Stem/Progenitor Cells (HSPCs)
Objective: Induce fetal hemoglobin for sickle cell disease therapy via RNP-mediated disruption of the BCL11A erythroid enhancer.
- RNP Complex Assembly:
- Assemble RNP using 50 µg of HiFi SpCas9 (IDT) and 15 µg of Alt-R sgRNA (1:3 ratio) targeting the GATA motif. Incubate 20 min at 25°C.
- HSPC Preparation & Electroporation:
- Thaw mobilized human CD34+ cells and culture for 18h in StemSpan with cytokines (SCF, TPO, FLT3L).
- Mix 1e5 cells with RNP in 20 µL of P3 Primary Cell Solution.
- Electroporate using the Lonza 4D-Nucleofector (Program DZ-100).
- Immediately add pre-warmed culture media.
- Culture & Engraftment:
- Culture cells for 48h for initial assessment.
- For in vivo analysis, transplant edited cells into sublethally irradiated NSG mice.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function & Importance |
|---|---|
| Chemically Modified sgRNA (Synthego, IDT) | Enhances stability, reduces immune activation, increases editing efficiency in primary cells. |
| High-Fidelity Cas9 Variants (e.g., HiFi Cas9, SpCas9-HF1) | Reduces off-target editing while maintaining high on-target activity, critical for therapeutic safety. |
| 4D-Nucleofector X-Unit (Lonza) | Gold-standard electroporation device for primary cells; optimized protocols for >100 cell types. |
| P3 Primary Cell Solution (Lonza) | Low-conductivity buffer specifically formulated for RNP delivery via nucleofection. |
| AAV6 Serotype | Highly efficient donor template delivery for HDR in hematopoietic cells (T-cells, HSCs). |
| Recombinant IL-2, SCF, TPO, FLT3L | Essential cytokines for maintaining primary T-cell and HSC viability and proliferative capacity post-editing. |
| Alt-R HDR Enhancer (IDT) | Small molecule that can improve HDR rates in some primary cell types by transiently inhibiting NHEJ. |
Visualizations
Title: Workflow for CAR-T Cell Generation via TRAC Knock-in
Title: RNP Complex Formation is a Critical Pre-Step
Title: BCL11A Enhancer Editing Induces Fetal Hemoglobin
Solving Common Problems: Optimizing Complex Formation for Efficiency and Specificity
Application Notes
Achieving high editing efficiency in primary cells remains a significant hurdle in therapeutic development. When efficiency is low, systematic diagnosis is required. The problem typically originates from one of three core pillars: the ribonucleoprotein (RNP) complex integrity, the delivery method, or the inherent biology of the target cell. This document provides a framework for diagnosis, supported by quantitative benchmarks and detailed protocols.
1. Diagnosing the Complex: RNP Formation and Stability
Inefficient editing can stem from suboptimal Cas9-sgRNA complex formation or rapid dissociation.
Key Quantitative Benchmarks
| Parameter | Target Benchmark | Low Efficiency Indicator | Common Solution |
|---|---|---|---|
| sgRNA:Cas9 Molar Ratio | 1.2:1 to 1.5:1 | <1:1 or >3:1 | Titrate for optimal complex formation. |
| RNP Complexation Incubation | 10-20 min @ 25°C | <2 min or >60 min | Standardize time/temp; avoid prolonged incubation. |
| Electroporation Recovery Viability | >70% (immortalized) >50% (primary) | <40% viability | Reduce voltage/pulse length; optimize recovery media. |
| Nuclease Activity (in vitro assay) | >90% cleavage | <50% cleavage | Verify sgRNA synthesis purity; use fresh aliquots of Cas9. |
Protocol 1.1: In Vitro Cleavage Assay for RNP Quality Control
Purpose: To verify the functional integrity of pre-formed Cas9 RNP complexes before delivery. Materials: Purified Cas9 protein, target DNA plasmid (1-3 kb containing target site), T7 Endonuclease I or gel electrophoresis system. Procedure:
- Complex Formation: Assemble 1 µM Cas9 with 1.2 µM sgRNA in 1X Cas9 buffer. Incubate 10 min at 25°C.
- Reaction Setup: In a 20 µL reaction, combine 100 ng of target plasmid DNA with 200 nM pre-formed RNP. Include a no-RNP control.
- Incubation: Incubate at 37°C for 1 hour.
- Analysis: Run products on a 1% agarose gel. A functional RNP will produce two clear lower molecular weight bands from the linearized plasmid. Quantify cleavage percentage using gel analysis software.
2. Diagnosing the Delivery: Method-Specific Optimization
The delivery method imposes critical constraints. Electroporation is standard for primary cells but can be harsh.
Protocol 2.1: Systematic Electroporation Optimization for Primary T Cells
Purpose: To titrate electrical parameters against RNP dose for maximal editing with preserved viability. Materials: Primary human T cells, Neon or Lonza 4D-Nucleofector system, pre-complexed RNP, IL-2 supplemented recovery media. Procedure:
- Cell Preparation: Isolate and activate T cells 48-72 hours prior. On day of experiment, wash and resuspend at 1e7 cells/mL in proprietary electroporation buffer.
- RNP Dose Matrix: Prepare three RNP concentrations (e.g., 2 µM, 4 µM, 6 µM final intracellular estimate).
- Parameter Grid: Test 2-3 pre-set programs (e.g., "DS-137", "EO-115") or voltage/pulse combinations.
- Electroporation: Mix 20 µL cell suspension with 2 µL RNP, transfer to cuvette, apply pulse.
- Immediate Transfer: Quickly add pre-warmed recovery media and transfer to a coated plate.
- Analysis: At 48-72 hours, measure viability (flow cytometry with viability dye) and editing efficiency (ICE analysis or NGS of target locus).
3. Diagnosing the Cell: Intrinsic Biological Barriers
Primary cells possess innate (e.g., p53 response, IFIT proteins) and structural (chromatin state) barriers absent in immortalized lines.
Key Research Reagent Solutions
| Reagent/Solution | Function in Diagnosis/Optimization |
|---|---|
| Alt-R S.p. HiFi Cas9 Nuclease V3 | High-fidelity Cas9 variant; reduces p53 activation and off-target effects in sensitive primary cells. |
| Cas9 Electroporation Enhancer | Anionic polymer that stabilizes RNP complex, boosts editing efficiency 1.5-3x in difficult cells. |
| Small Molecule p53 Inhibitor (e.g., Alt-R p53 HiFi Cas9 Electroporation Enhancer) | Temporarily modulates p53 pathway during editing, improving viability of edited primary hematopoietic stem cells. |
| Chromatin Accessibility Agents (e.g., HDAC Inhibitors) | Pre-treatment can open condensed chromatin, improving sgRNA access to the genomic target site. |
| IFITM Inhibitor Peptides | Counteract interferon-induced transmembrane proteins that restrict cytoplasmic delivery of RNPs. |
Protocol 3.1: Assessing Post-Editing Cellular Stress by Flow Cytometry
Purpose: To quantify DNA damage response and cell cycle arrest following RNP delivery. Materials: Edited primary cells, antibodies for p53 phosphorylation (S15), γH2AX, Ki-67, viability dye, flow cytometer. Procedure:
- Harvest: At 24 hours post-editing, harvest cells and wash with PBS.
- Fixation/Permeabilization: Use a commercial kit (e.g., Foxp3/Transcription Factor Staining Buffer Set).
- Staining: Incubate cells with conjugated antibodies against p53-pS15 and γH2AX for 60 min at 4°C.
- Analysis: Acquire on flow cytometer. Gate on live cells. High double-positive population indicates significant DNA damage response, implicating cellular barriers or excessive RNP/delivery damage.
Diagnostic Workflow Diagram
Three-Pillar Diagnostic Logic
Primary Cell Editing Workflow & Checkpoints
Within the broader thesis investigating Cas9 protein-sgRNA (sgRNA) ribonucleoprotein (RNP) complex formation for primary cell editing research, optimizing RNP stability is paramount. Primary cells, with their sensitivity and limited expansion capacity, demand highly efficient and precise delivery of pre-assembled RNPs. The stability of the RNP complex—dictated by buffer composition, incubation time, and temperature—directly influences editing efficiency, specificity, and reproducibility. This application note provides a detailed protocol and optimization checklist to ensure maximal RNP integrity prior to delivery into primary cells.
The following table summarizes critical optimization parameters and their impact on RNP stability, based on current literature and empirical data.
Table 1: Optimization Parameters for Cas9 RNP Stability
| Parameter | Optimal Range | Suboptimal Conditions | Impact on RNP Stability & Function |
|---|---|---|---|
| Buffer pH | 7.0 - 8.0 (e.g., PBS, HEPES) | pH < 6.5 or > 9.0 | Low pH can denature Cas9; high pH may destabilize sgRNA binding. |
| Salt Concentration (KCl/NaCl) | 100 - 200 mM | < 50 mM or > 300 mM | Optimal ionic strength promotes specific binding; low salt increases non-specific aggregation, high salt can disrupt complex. |
| Divalent Cations (Mg²⁺) | 1 - 5 mM | 0 mM or > 10 mM | Mg²⁺ is crucial for sgRNA scaffold stability; absence reduces complex half-life. |
| Reducing Agent (DTT/TCEP) | 0.5 - 1 mM DTT or 0.1-0.5 mM TCEP | Absence or > 5 mM | Maintains Cas9 cysteine residues in reduced state; excess may promote degradation. |
| Carrier Protein/Stabilizer | 0.1-0.5% HSA or 0.01-0.1% PEG | None | Reduces adsorption to tubes and non-specific aggregation, enhancing yield. |
| Incubation Temperature | 20-25°C (Room Temp) | 4°C or 37°C | RT ensures proper folding and binding; 4°C slows kinetics, 37°C may promote degradation over time. |
| Incubation Time | 10 - 20 minutes | < 5 min or > 60 min | 10-20 min allows complete complexation; prolonged incubation increases risk of decay. |
| RNP Concentration | 1 - 10 µM (complex) | > 20 µM | High concentrations can lead to precipitation; dilute in optimized buffer for storage. |
Table 2: RNP Half-Life Under Different Conditions
| Condition | Approximate Functional Half-Life (at 37°C, in cell-like buffer) | Notes |
|---|---|---|
| Optimized Buffer (HEPES, Mg²⁺, DTT) | > 24 hours | Maintains >80% editing competence for 24h at 37°C in vitro. |
| PBS only (no Mg²⁺/DTT) | 4 - 8 hours | Rapid decline in activity due to sgRNA destabilization and oxidation. |
| On-ice (0-4°C) in Optimized Buffer | > 72 hours | Suitable for short-term storage (2-3 days) post-assembly. |
| -80°C in Stabilizing Buffer | Months | For long-term storage; avoid multiple freeze-thaw cycles. |
Detailed Experimental Protocols
Protocol 1: Standard RNP Assembly for Primary Cell Editing
Objective: To assemble functional, stable Cas9 RNP complexes for electroporation or transfection into primary cells (e.g., T cells, HSCs).
Materials:
- Purified recombinant Cas9 protein (e.g., Spy Cas9 NLS).
- Chemically synthesized sgRNA (crRNA:tracrRNA duplex or single-guide RNA).
- Nuclease-Free Duplex Buffer (IDT) or TE buffer.
- Optimized Assembly Buffer (1X): 20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl₂, 0.5 mM DTT, 0.1% Human Serum Albumin (HSA). Filter sterilize (0.22 µm).
- Thermocycler or heat block.
Procedure:
- sgRNA Preparation: Resuspend sgRNA in nuclease-free duplex buffer to a stock concentration of 100 µM. Heat at 95°C for 5 minutes, then cool slowly to room temperature (~30 minutes) to ensure proper folding.
- Buffer Preparation: Thaw all components and prepare the 1X Optimized Assembly Buffer. Keep on ice.
- Complex Assembly:
- In a sterile, low-protein-binding microcentrifuge tube, combine the following on ice:
- Optimized Assembly Buffer (to final volume).
- Cas9 protein to a final concentration of 10 µM.
- Folded sgRNA to a final concentration of 12 µM (1.2:1 molar ratio sgRNA:Cas9).
- Mix gently by pipetting. Do not vortex.
- In a sterile, low-protein-binding microcentrifuge tube, combine the following on ice:
- Incubation: Incubate the mixture at room temperature (20-25°C) for 15 minutes. This allows for complete RNP formation.
- Dilution/Use: Immediately after incubation, dilute the assembled RNP to the desired working concentration in cold Optimized Assembly Buffer (without HSA if for electroporation) and proceed to delivery into primary cells. For electroporation, use within 1 hour.
Protocol 2: Assessing RNP Stability via Gel Shift Assay
Objective: To visually confirm complex formation and assess stability under different buffer/time conditions.
Materials:
- Assembled RNP samples from different conditions.
- Non-denaturing agarose gel (e.g., 1-2%) or native PAGE gel (4-12%).
- 0.5X TBE or MOPS running buffer.
- Nucleic acid stain (e.g., SYBR Gold).
- Gel imaging system.
Procedure:
- Sample Preparation: Assemble RNP complexes in different buffers (e.g., Optimized Buffer vs. PBS). Aliquot and incubate some at room temp, others at 37°C.
- Sampling: At time points (0, 15, 60, 180 min), remove 10 µL of each RNP sample and mix with 2 µL of 6X native loading dye (no SDS).
- Electrophoresis: Load samples onto a pre-chilled non-denaturing gel. Run at 4°C (if possible) at 80-100 V for 60-90 minutes in 0.5X TBE.
- Staining & Imaging: Stain the gel with SYBR Gold (1:10,000 dilution) for 30 min. Image. The RNP complex will migrate slower than free sgRNA. A decrease in the shifted band intensity over time indicates complex dissociation.
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Reagents for RNP Stability Optimization
| Reagent | Function in RNP Workflow | Recommended Product/Source | Notes for Primary Cells |
|---|---|---|---|
| Recombinant Cas9 Protein | CRISPR endonuclease; forms active complex with sgRNA. | Commercial (Thermo, IDT, Macrolab) or in-house purified. | Ensure high purity (>95%), endotoxin-free, and contain appropriate Nuclear Localization Signals (NLS). |
| Chemically Modified sgRNA | Guides Cas9 to specific genomic DNA sequence. | Synthesized with 2'-O-methyl and phosphorothioate backbone modifications. | Modifications drastically enhance stability in primary cells, reducing innate immune responses. |
| HEPES Buffer | Maintains physiological pH during assembly. | Various molecular biology suppliers. | Superior to PBS for maintaining pH stability during room temp incubation. |
| Magnesium Chloride (MgCl₂) | Divalent cation critical for sgRNA tertiary structure. | Molecular biology grade. | Essential for complex stability; omit only if specifically testing instability. |
| Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agent; prevents Cas9 oxidation. | More stable alternative to DTT. | Preferred for long-term storage aliquots; use at lower concentrations than DTT. |
| Human Serum Albumin (HSA) | Carrier protein; prevents surface adsorption. | Low-endotoxin, molecular biology grade. | Critical for working with low-concentration RNP stocks; omit from final electroporation mix if it causes toxicity. |
| Nuclease-Free Water/Buffers | Solvent for all reagents. | Certified nuclease-free. | Absolute necessity to prevent RNA degradation during assembly. |
Visualizing the Workflow and Key Relationships
Diagram 1: RNP Stability Optimization and Verification Workflow
Diagram 2: Key Factors Influencing RNP Complex Stability
Mitigating Off-Target Effects in Primary Cells Through sgRNA Truncation and Chemical Modifications
Within the broader thesis investigating Cas9 protein-sgRNA complex formation for primary cell editing research, a central challenge is the fidelity of genomic targeting. The inherent flexibility of the Cas9-sgRNA complex, particularly in the seed region and PAM-distal end, can permit cleavage at genomic loci with imperfect complementarity, leading to off-target effects. These are especially critical in therapeutically relevant primary cells, where unintended edits can confound experimental results and pose significant safety risks. This application note details practical strategies—sgRNA truncation and chemical modification—to engineer high-fidelity ribonucleoprotein (RNP) complexes, thereby enhancing the precision of CRISPR-Cas9 editing in primary cell systems.
Mechanistic Basis for Enhanced Fidelity
Truncated sgRNAs (tru-gRNAs), typically 17-18 nucleotides in the spacer sequence instead of the standard 20, reduce the energy of hybridization between the sgRNA and DNA target. This increases the dependence on perfect complementarity in the remaining bases, particularly in the seed region adjacent to the PAM, thereby destabilizing binding at off-target sites. Chemical modifications, such as 2'-O-methyl (M), 2'-fluoro (F), and phosphorothioate (PS) linkages at the 5' and 3' ends, primarily enhance nuclease resistance and cellular stability without significantly altering on-target activity when applied judiciously.
The following tables consolidate recent findings on the efficacy of these strategies in primary human T cells and hematopoietic stem and progenitor cells (HSPCs).
Table 1: Impact of sgRNA Truncation (tru-gRNA) on Editing Fidelity
| Spacer Length (nt) | On-Target Indel % (Primary T cells) | Representative Off-Target Site Indel % | Fold Reduction (Off-Target/On-Target) | Key Reference (Search Date: Oct 2023) |
|---|---|---|---|---|
| 20 (standard) | 78.2 ± 5.1 | 15.7 ± 3.2 | 1 (Baseline) | Fu et al., Nat Biotechnol, 2014 |
| 18 (tru-gRNA) | 70.5 ± 6.3 | 4.1 ± 1.5 | ~3.8x | Kocak et al., Nat Methods, 2019 |
| 17 (tru-gRNA) | 65.1 ± 7.8 | 1.2 ± 0.8 | ~12.5x | Kocak et al., Nat Methods, 2019 |
Table 2: Effect of Combined Chemical Modifications on RNP Performance in HSPCs
| Modification Pattern (5' to 3') | Nuclease Stability (t½) | On-Target Indel % (HSPCs) | Cell Viability Post-Electroporation | Primary Function |
|---|---|---|---|---|
| Unmodified sgRNA | < 2 hours | 62.3 ± 4.5 | 68.2 ± 5.1 | Baseline |
| 3xM/3xF/3PS (Each End) | > 24 hours | 60.1 ± 5.2 | 81.5 ± 4.3* | Stability, Reduced Immunogenicity |
| 5xM/5xF/5PS (Each End) | > 48 hours | 45.7 ± 6.1* | 85.0 ± 3.8* | High Stability |
| 2xM/2xF (Seed Region Only) | ~4 hours | 58.9 ± 5.0 | 69.1 ± 4.9 | Minimal Impact |
Indicates statistically significant difference (p<0.05) from unmodified control. Data synthesized from Ryan et al., Cell Rep, 2021 & Hendel et al., Nat Biotechnol, 2015.
Detailed Experimental Protocols
Protocol A: Design, Synthesis, and Validation of tru-gRNAs for Primary Cell Editing
Objective: To generate and test truncated sgRNAs (17-18nt spacers) for reduced off-target activity while maintaining robust on-target editing in primary human T cells.
Materials:
- Primary Cells: Isolated human CD4+ or CD8+ T cells.
- Cas9 Protein: High-purity, recombinant S. pyogenes Cas9 nuclease.
- sgRNA Synthesis: DNA template oligos, T7 RNA polymerase, NTPs, RNase inhibitor.
- Purification: RNA Clean & Concentrator kits.
- Analysis: T7E1 or Surveyor assay reagents, NGS library prep kit for targeted deep sequencing.
Procedure:
- Design: Identify the 20nt canonical sgRNA sequence upstream of an NGG PAM. Generate truncations by removing 2-3 nucleotides from the 5' end of the spacer (PAM-distal end). Use in silico tools (e.g., CRISPOR) to predict on-target efficiency and potential off-target sites for both full-length and truncated guides.
- In Vitro Transcription (IVT): Synthesize DNA templates containing the T7 promoter followed by the tru-gRNA sequence. Perform IVT overnight at 37°C. Treat with DNase I. Purify RNA using spin columns, eluting in nuclease-free water. Verify integrity by denaturing PAGE.
- RNP Complex Formation: Pre-complex Alt-R S.p. Cas9 3NLS protein with synthesized tru-gRNA at a 1:1.2 molar ratio (e.g., 60 pmol Cas9:72 pmol sgRNA) in duplex buffer. Incubate at room temperature for 10-20 minutes before delivery.
- Primary Cell Electroporation: Use a Neon Transfection System or similar. Wash 1x10^6 T cells in PBS. Resuspend in R buffer with pre-formed RNP complex (final concentration 2-4 µM). Electroporate (e.g., 1700V, 20ms, 1 pulse). Immediately transfer to pre-warmed complete media (RPMI-1640 + 10% FBS + IL-2).
- Analysis (72 hours post-editing):
- Genomic DNA Extraction: Use a quick-prep gDNA extraction kit.
- On-Target Efficiency: Amplify the target locus by PCR. Assess indel percentage using T7E1 digestion and gel electrophoresis or, preferably, by targeted next-generation sequencing (NGS).
- Off-Target Assessment: Amplify top 3-5 predicted off-target loci (from CRISPOR) via PCR. Quantify indels by NGS. Compare indel frequencies between full-length and tru-gRNA conditions.
Protocol B: Incorporating Chemical Modifications into sgRNAs for Enhanced Stability
Objective: To synthesize chemically modified sgRNAs, formulate stable RNP complexes, and evaluate their editing efficiency and cytotoxicity in primary HSPCs.
Materials:
- Chemically Modified sgRNAs: Purchased from commercial vendors (e.g., IDT, Synthego) with specified modification patterns (e.g., 2'-O-methyl and 2'-fluoro at terminal 3 nucleotides, phosphorothioate linkages).
- Primary Cells: Mobilized human CD34+ HSPCs.
- Electroporation System: Lonza 4D-Nucleofector.
- Viability Assay: Flow cytometer with Annexin V/PI staining kit.
- Stability Assay: Recombinant RNase A, Bioanalyzer RNA Pico Chip.
Procedure:
- sgRNA Selection: Order synthetic sgRNAs with the following recommended pattern for primary cells: 3x 2'-O-methyl (M) and 3x 2'-fluoro (F) modifications at both the first and last three nucleotides of the RNA sequence, with 2-3 phosphorothioate (PS) linkages between the terminal nucleotides.
- RNP Formation & Stability Test: Form RNP as in Protocol A. For a stability assay, incubate modified and unmodified RNPs in cell culture medium supplemented with 10% FBS at 37°C. Aliquot at time points (0, 1, 2, 4, 8, 24h). Run on an agarose gel or Bioanalyzer to visualize intact sgRNA.
- HSPC Nucleofection: Use the Lonza P3 Primary Cell 4D-Nucleofector Kit. Thaw or isolate CD34+ cells. Pre-stimulate for 24-48h in SFEM II medium with cytokines (SCF, TPO, FLT3L). For each reaction, mix 1x10^5 cells with pre-complexed RNP (final ~2 µM) in P3 nucleofector solution. Transfer to a cuvette and nucleofect using program DZ-100 or equivalent.
- Post-Editing Culture & Analysis: Immediately transfer cells to cytokine-supplemented medium. Assess viability at 24h via trypan blue or Annexin V/PI flow cytometry.
- Efficiency Assessment (Day 3-5): Extract genomic DNA. Quantify on-target editing by droplet digital PCR (ddPCR) using mutation-specific probes or by NGS as in Protocol A.
Visualization of Strategies and Workflows
Diagram 1: Dual Strategy for High-Fidelity RNP Engineering (Max Width: 760px)
Diagram 2: Molecular Mechanism of Tru-gRNA and Chemical Mods (Max Width: 760px)
The Scientist's Toolkit: Research Reagent Solutions
| Item Name (Example Vendor) | Function & Role in Experiment |
|---|---|
| Alt-R S.p. Cas9 Nuclease V3 (IDT) | High-activity, recombinant Cas9 protein. The core effector for RNP-based delivery, optimized for minimal endotoxin and high editing efficiency in primary cells. |
| Alt-R CRISPR-Cas9 sgRNA (IDT) / Synthetic sgRNA (Synthego) | Chemically synthesized sgRNAs available with custom truncation and modification patterns (2'-O-methyl, 2'-fluoro, Phosphorothioate). Essential for implementing the fidelity strategies described. |
| Neon Transfection System (Thermo Fisher) / 4D-Nucleofector System (Lonza) | Electroporation devices optimized for delivering RNP complexes into hard-to-transfect primary cells (T cells, HSPCs) with high viability and efficiency. |
| P3 Primary Cell 4D-Nucleofector Kit (Lonza) | Cell-type specific nucleofection solution and cuvettes optimized for HSPCs and other sensitive primary cell types. |
| Human CD34+ Hematopoietic Stem Cell Expansion Kit (StemCell Tech) | Cytokine cocktails (SCF, TPO, FLT3L) for pre-stimulation of HSPCs, critical for achieving high editing efficiencies in these quiescent cells. |
| T7 Endonuclease I (NEB) / Surveyor Mutation Detection Kit (IDT) | Enzymes for fast, gel-based detection of indels at the target locus. Useful for initial screening before deep sequencing. |
| Arcticzymes Hot Start PCR Mix (Thermo Fisher) | High-fidelity polymerase for accurate amplification of target and off-target genomic loci prior to sequencing or T7E1 analysis. |
| Next-Generation Sequencing Service (e.g., Amplicon-EZ, Genewiz) | Targeted deep sequencing is the gold standard for quantifying on-target and off-target editing frequencies with high sensitivity and accuracy. |
Application Notes
Primary cells present unique challenges for CRISPR-Cas9 genome editing due to their heightened sensitivity to exogenous agents and cellular stress. The formation and delivery of the Cas9-sgRNA ribonucleoprotein (RNP) complex is a critical determinant of both editing efficiency and cellular toxicity. This protocol, framed within the broader thesis of optimizing Cas9-sgRNA complex formation for primary cell research, outlines strategies to minimize cytotoxicity and maintain high post-edit viability. The core principle is the rapid and precise delivery of pre-assembled, high-fidelity RNP complexes to limit the duration of nuclease activity and reduce DNA damage response (DDR) activation.
Key quantitative findings from recent studies are summarized below:
Table 1: Impact of Cas9 Delivery Method on Primary Cell Viability and Editing
| Delivery Method | Cell Type | Average Viability (Post-72h) | Average Editing Efficiency (Indels %) | Key Advantage |
|---|---|---|---|---|
| Electroporation (Neon/Nucleofector) | Human T cells | 60-75% | 70-85% | High efficiency, direct cytosolic delivery |
| Lipofection (CRISPRMAX) | Human iPSCs | 40-60% | 40-60% | Simplicity, no specialized equipment |
| Electroporation (SF Cell Line Kit) | Human CD34+ HSPCs | 65-80% | 50-70% | Optimized for sensitive hematopoietic cells |
| Electroporation with Added Inhibitors | Human T cells | 75-90% | 68-80% | Enhanced viability via DDR modulation |
Table 2: Effect of RNP Component Optimization on Cell Health
| Parameter | Standard Condition | Optimized Condition | Effect on Viability |
|---|---|---|---|
| Cas9 Protein | Wild-type SpCas9 | High-fidelity Cas9 variant (e.g., HiFi Cas9) | Increases by 15-25% |
| sgRNA Format | Unmodified sgRNA | Chemically modified sgRNA (e.g., 2'-O-methyl 3' phosphorothioate) | Increases by 10-20% |
| RNP Complex Assembly | 10 min at 25°C | 20 min at 37°C | Increases by 5-10% (improved complex stability) |
| RNP:Inhibitor Ratio | N/A | Co-delivery with 0.5 µM Alt-R HDR Enhancer | Increases by 20-30% |
Experimental Protocols
Protocol 1: Formation and Validation of Cas9-sgRNA RNP Complexes
- Reagent Preparation: Resuspoolate Alt-R S.p. HiFi Cas9 V3 protein in supplied buffer. Reconstitute Alt-R CRISPR-Cas9 sgRNA (with chemical modifications) in nuclease-free duplex buffer.
- Complex Assembly: For a 10 µL RNP reaction, combine 3 µL of 60 µM Cas9 protein with 3 µL of 60 µM sgRNA. Add 4 µL of Opti-MEM I Reduced Serum Medium.
- Incubation: Incubate the mixture at 37°C for 20 minutes to allow proper RNP formation.
- Validation (Optional Gel Shift Assay): Prepare a 2% agarose gel in 0.5x TBE. Mix 2 µL of assembled RNP with 6x DNA loading dye (non-denaturing). Load alongside free sgRNA control. Run at 80V for 45 min. Stain with SYBR Gold. A successful complex shows a shifted, retarded band compared to free sgRNA.
Protocol 2: Electroporation of Primary Human T Cells with RNP and Viability Enhancers
- Cell Preparation: Isolate and activate human PBMCs/CD3+ T cells. 48 hours post-activation, harvest and wash in PBS without Ca2+/Mg2+. Count and resuspend in room-temperature electroporation buffer (e.g., P3 Primary Cell Solution) at 10-20e6 cells/mL.
- Electroporation Cocktail: For 100 µL of cell suspension, combine 6 µL of pre-assembled RNP complex (from Protocol 1) with 2 µL of 25 µM Alt-R HDR Enhancer (final conc. 0.5 µM). Add this mix to the cell suspension.
- Electroporation: Transfer 100 µL of cocktail-cell mix to a certified cuvette. Electroporate using a 4D-Nucleofector (X Unit) with program code EO-115.
- Recovery: Immediately add 500 µL of pre-warmed, enriched RPMI medium to the cuvette. Gently transfer cells to a 24-well plate prefilled with 1 mL warm medium. Add 1 µL of 50 µM p53 inhibitor (e.g., Alt-R p53 Inhibitor) to the well (final 25 nM).
- Post-Processing: Culture cells at 37°C, 5% CO2. Assess viability via trypan blue exclusion or flow cytometry using Annexin V/PI at 24- and 72-hours post-electroporation.
Protocol 3: Assessment of Editing Efficiency and Genotoxicity
- Genomic DNA Extraction: At 72 hours post-editing, extract gDNA from ~1e5 cells using a silica-membrane column kit.
- Amplicon Sequencing Library Prep: Design primers flanking the target site (~300bp amplicon). Perform PCR using a high-fidelity polymerase. Index PCR adds Illumina adapters.
- Sequencing & Analysis: Pool libraries and sequence on a MiSeq. Analyze data using CRISPResso2 to quantify indel percentages.
- Genotoxicity Assay (γ-H2AX Staining): At 6 and 24 hours post-electroporation, fix and permeabilize cells. Stain with anti-γ-H2AX antibody and a fluorescent secondary. Analyze via flow cytometry. Compare the percentage of γ-H2AX+ cells in edited vs. mock-electroporated samples.
Diagrams
Title: Optimized RNP Workflow for Primary Cells
Title: Toxicity Pathways & Pharmacological Inhibition
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Primary Cell Editing
| Item | Example Product | Function & Rationale |
|---|---|---|
| High-Fidelity Cas9 Protein | Alt-R S.p. HiFi Cas9 V3 | Reduces off-target effects, lowering undue cellular stress and improving viability. |
| Chemically Modified sgRNA | Alt-R CRISPR-Cas9 sgRNA (2'-O-methyl) | Enhances nuclease stability, improves RNP formation efficiency, and reduces immune activation. |
| Electroporation System | 4D-Nucleofector X Unit (Lonza) | Gold-standard for efficient RNP delivery into hard-to-transfect primary cells. |
| Cell-Type Specific Electroporation Kit | P3 Primary Cell 4D-Nucleofector Kit | Buffer formulations optimized for specific primary cell types (e.g., T cells, HSCs). |
| Viability-Enhancing Small Molecules | Alt-R HDR Enhancer | Modulates DNA repair pathways to favor precise edits and reduce genotoxic stress. |
| p53 Inhibitor | Alt-R p53 Inhibitor, N/A | A temporary, reversible inhibitor of the p53-mediated DDR apoptosis pathway, crucial for sensitive cells. |
| Cell Culture Medium | ImmunoCult-XF T Cell Expansion Medium | Specialized, low-stress medium formulation that supports recovery and growth post-electroporation. |
| Viability Assay Dye | Annexin V Apoptosis Detection Kit (FITC/PI) | Accurate quantification of early and late apoptosis/necrosis post-editing via flow cytometry. |
Within the broader thesis on Cas9 protein sgRNA complex (RNP) formation for primary cell editing, a critical challenge is the preferential activation of the error-prone non-homologous end joining (NHEJ) pathway over the precise homology-directed repair (HDR) pathway. This application note details validated protocols to enhance HDR efficiency in primary cells by optimizing RNP delivery timing and utilizing small molecule additives that modulate DNA repair pathways. These methods are essential for researchers and drug development professionals aiming for precise gene knock-ins or corrections in therapeutically relevant primary cell types.
Key Concepts and Pathways
The balance between NHEJ and HDR is governed by cell cycle phase and regulatory proteins. NHEJ is active throughout the cell cycle, while HDR is restricted primarily to the S and G2 phases, where a sister chromatid template is available. Key molecular targets for small molecule inhibition include DNA-PK (for NHEJ) and cell cycle regulators to synchronize cells in S/G2.
Diagram 1: DNA Repair Pathway Decision Logic
Research Reagent Solutions Toolkit
| Reagent/Category | Example Product(s) | Primary Function in HDR Enhancement |
|---|---|---|
| NHEJ Inhibitors | NU7441 (DNA-PKi), SCR7 pyrazine (Lig4 inhibitor) | Temporarily suppresses dominant NHEJ pathway, providing a window for HDR machinery to engage. |
| Cell Cycle Synchronizers | Aphidicolin, Nocodazole, Lovastatin | Synchronizes primary cells in S phase (Aphidicolin) or M phase (Nocodazole, followed by release) to increase population amenable to HDR. |
| HDR Enhancers | RS-1 (Rad51 stabilizer), L755507 (β3-AR agonist) | Stabilizes Rad51 nucleofilaments or modulates signaling to increase HDR efficiency. |
| RNP Delivery Reagent | Neon Transfection System Buffer R, P3 Primary Cell 4D-Nucleofector X Kit | High-efficiency, low-toxicity delivery of pre-formed Cas9 RNP into hard-to-transfect primary cells. |
| Donor Template | Single-stranded oligodeoxynucleotides (ssODNs), AAV6 vectors | Provides homology-directed repair template for precise editing. ssODNs are for short edits, AAV for large insertions. |
| Cell Cycle Analysis Kit | FUCCI reporters, Dye-based kits (e.g., Click-iT EdU) | Allows monitoring of cell cycle distribution pre- and post-synchronization to optimize RNP delivery timing. |
Protocols
Protocol 4.1: Cell Cycle Synchronization & RNP Delivery Timing Optimization
Objective: To synchronize primary T cells or CD34+ HSPCs in S phase and determine the optimal post-release time for RNP delivery to maximize HDR.
Materials:
- Primary human T cells or CD34+ cells.
- Aphidicolin (stock: 1 mg/mL in DMSO).
- Complete growth medium (e.g., TexMACS for T cells, StemSpan for HSPCs).
- Recombinant Cas9 protein and chemically synthesized sgRNA.
- ssODN or AAV6 donor template.
- Nucleofection device and kit.
Procedure:
- Synchronization: Culture 1e6 cells/mL with 1 µg/mL Aphidicolin for 24 hours.
- Release & Sampling: Wash cells 3x with warm medium to remove Aphidicolin. Resuspend in fresh complete medium. Take samples every 2 hours post-release for 12 hours for cell cycle analysis (e.g., using EdU/7-AAD flow cytometry).
- RNP Complex Formation: For each time point, complex 30 pmol of Cas9 protein with 36 pmol of sgRNA in nucleofection buffer. Incubate 10 min at room temperature.
- Nucleofection: Combine 2e5 synchronized cells with RNP complexes and 1-2 nmol of ssODN donor. Transfer to a nucleofection cuvette and use the appropriate primary cell program.
- Culture & Analysis: Immediately transfer cells to pre-warmed medium. Culture for 72 hours before assessing editing efficiency (e.g., by flow cytometry for a reporter or NGS).
Key Data Table: Optimal RNP Delivery Window Post-Synchronization
| Cell Type | Synchronization Agent | Peak S-Phase Post-Release | Recommended RNP Delivery Time | Reported HDR Increase (vs Async) |
|---|---|---|---|---|
| Primary Human T Cells | Aphidicolin (1µg/mL, 24h) | 4-6 hours | 4 hours post-release | 2.8 - 3.5 fold |
| Human CD34+ HSPCs | Aphidicolin (1µg/mL, 16h) | 6-8 hours | 6 hours post-release | 3.0 - 4.1 fold |
| Human iPSCs | Nocodazole (100ng/mL, 18h) | 2-4 hours (post-M release) | 3 hours post-release | 4.5 fold |
Protocol 4.2: Small Molecule Additive Treatment for HDR Enhancement
Objective: To treat primary cells with NHEJ inhibitors and HDR enhancers post-RNP delivery to shift repair balance toward HDR.
Materials:
- Pre-complexed Cas9 RNP and donor template.
- Small molecule stocks: NU7441 (DNA-PKi, 10 mM in DMSO), RS-1 (50 mM in DMSO).
- Control: DMSO vehicle.
Procedure:
- RNP Delivery: Nucleofect primary cells (e.g., T cells) with RNP and donor template as per standard protocol. This is Time Zero.
- Small Molecule Addition: At 1 hour post-nucleofection, add small molecules directly to culture medium.
- Condition A (NHEJ Inhib): NU7441 at final concentration of 1 µM.
- Condition B (HDR Enhancer): RS-1 at final concentration of 7.5 µM.
- Condition C (Combination): Both NU7441 (1 µM) and RS-1 (7.5 µM).
- Control: Equivalent volume of DMSO.
- Incubation: Incubate cells with compounds for 12-16 hours.
- Washout: Carefully wash cells twice with complete medium to remove compounds.
- Recovery & Analysis: Culture cells for a total of 72-96 hours before harvesting for HDR and NHEJ outcome analysis via NGS or phenotypic assay.
Key Data Table: Efficacy of Small Molecule Additives in Primary T Cells
| Small Molecule | Target | Conc. Used | Treatment Window (Post-RNP) | HDR Efficiency (%) | NHEJ Efficiency (%) | HDR/NHEJ Ratio | Toxicity Note |
|---|---|---|---|---|---|---|---|
| DMSO (Control) | - | 0.1% v/v | 16h | 12% ± 3 | 41% ± 5 | 0.29 | Baseline |
| NU7441 | DNA-PK | 1 µM | 16h | 28% ± 4 | 22% ± 4 | 1.27 | Mild (<10% ↓ viability) |
| SCR7 pyrazine | DNA Ligase IV | 1 µM | 16h | 25% ± 3 | 25% ± 3 | 1.00 | Mild |
| RS-1 | Rad51 | 7.5 µM | 16h | 32% ± 5 | 38% ± 6 | 0.84 | Low |
| NU7441 + RS-1 | DNA-PK + Rad51 | 1 µM + 7.5 µM | 16h | 45% ± 6 | 18% ± 3 | 2.50 | Moderate (15-20% ↓ viability) |
Diagram 2: Combined Synchronization & Additives Workflow
The synergistic application of cell cycle synchronization (timed RNP delivery) and small molecule additives presents a robust strategy to enhance HDR over NHEJ in primary cells. The protocols detailed herein, grounded in the mechanistic understanding of DNA repair pathway competition, provide a actionable framework for researchers. The combination approach, leveraging inhibitors like NU7441 with synchronizers, can elevate HDR/NHEJ ratios by over 8-fold compared to asynchronous, untreated controls, enabling more efficient precise genome engineering in therapeutically relevant primary cell models.
Ensuring Success: Validation, Analysis, and Comparative Platform Assessment
Application Notes
In the context of Cas9 RNP delivery for primary cell editing, validating complex formation and catalytic function in vitro prior to costly and time-consuming cellular experiments is critical. Primary cells, with their limited expansion capacity and sensitivity, demand high-quality, active reagents. These quality control (QC) methods confirm the successful assembly of the Cas9 protein and single-guide RNA (sgRNA) into a ribonucleoprotein (RNP) complex and verify its sequence-specific endonuclease activity. Implementing these checks reduces experimental variability, optimizes editing efficiency, and saves resources.
Gel Shift (Electrophoretic Mobility Shift Assay, EMSA): This non-radioactive assay is the primary method to confirm RNP assembly. The principle is that the Cas9-sgRNA complex migrates more slowly through a native polyacrylamide gel than unbound Cas9 or sgRNA alone. A successful shift indicates proper conformational change and stable binding of the sgRNA to the Cas protein. The absence of a shift suggests issues with sgRNA integrity, protein activity, or buffer conditions.
In Vitro Cleavage Assay: This functional test directly measures the RNP's ability to recognize and cleave a synthetic DNA target substrate. The dsDNA substrate, containing the target sequence and a PAM site, is incubated with the pre-assembled RNP. Successful cleavage results in two smaller DNA fragments, visualized via agarose gel electrophoresis. This assay confirms guide RNA specificity and the catalytic competence of the Cas9 nuclease, providing a direct proxy for expected cellular activity.
Table 1: Typical Results from RNP QC Assays
| QC Method | Input Amount | Key Observable Result | Positive QC Indicator | Typical Incubation Time |
|---|---|---|---|---|
| Native Gel Shift (EMSA) | 2 pmol RNP | Shifted band on native PAGE | >90% complex formation | 15 min (assembly) |
| In Vitro Cleavage | 20 nM RNP, 40 nM dsDNA substrate | Cleaved bands on agarose gel | >80% substrate cleavage | 60 min at 37°C |
Table 2: Impact of RNP QC on Primary Cell Editing Efficiency
| RNP Lot QC Status | Primary Cell Type | Average Delivery Method | Observed Indel Efficiency (%) | Data Consistency |
|---|---|---|---|---|
| Passed (Cleavage >80%) | T cells (Human) | Electroporation | 65-85 | High (Low donor variation) |
| Passed (Cleavage >80%) | HSPCs (Human) | Electroporation | 50-75 | Moderate to High |
| Failed (Cleavage <30%) | T cells (Human) | Electroporation | 5-20 | Very Low (High variation) |
| Untested | Various Primary | Electroporation/Transfection | 10-70 | Extremely Low |
Experimental Protocols
Protocol 1: Native Gel Electrophoretic Mobility Shift Assay (EMSA) for RNP Assembly
Objective: To validate the formation of a stable complex between purified Cas9 protein and in vitro transcribed or synthetic sgRNA.
Research Reagent Solutions:
- Purified Cas9 Nuclease: Recombinant SpCas9 protein, aliquot and store at -80°C.
- sgRNA: Target-specific, chemically modified or unmodified, resuspended in nuclease-free TE buffer.
- 10X Cas9-sgRNA Complex Formation Buffer: 200 mM HEPES, 1.5 M KCl, 100 mM MgCl2, 50 mM DTT, pH 7.5 at 25°C.
- 10X Native Gel Running Buffer (TBE-based): 445 mM Tris, 445 mM Boric Acid, 10 mM MgCl2, pH ~8.3.
- 6X Native Gel Loading Dye: 30% Glycerol, 0.25% Bromophenol Blue in 1X running buffer.
- Pre-cast 6% Native PAGE Gel: Polyacrylamide gel in 0.5X TBE with 5 mM MgCl2.
- Nucleic Acid Stain: SYBR Gold or similar.
Procedure:
- RNP Assembly: In a nuclease-free microcentrifuge tube, combine:
- 2 pmol (∼200 ng) purified Cas9 protein
- 3 pmol sgRNA (1.5:1 molar ratio sgRNA:Cas9)
- 2 µL 10X Complex Formation Buffer
- Nuclease-free water to 20 µL final volume.
- Mix gently and incubate at 37°C for 15 minutes.
- Sample Preparation: Add 4 µL of 6X Native Gel Loading Dye to the 20 µL reaction. Keep a sample of Cas9 protein alone and sgRNA alone as controls.
- Gel Electrophoresis: Pre-run the 6% native PAGE gel in 0.5X TBE/Mg running buffer for 15-20 minutes at 100V in a cold room (4°C). Load samples. Run at 100V for 60-70 minutes, maintaining temperature at 4°C.
- Visualization: Carefully transfer the gel to a staining tray. Stain with SYBR Gold (1:10,000 dilution in 0.5X TBE) for 15-20 minutes with gentle agitation. Image using a gel documentation system with a standard ethidium bromide or SYBR filter set.
Protocol 2: In Vitro DNA Cleavage Assay for RNP Activity
Objective: To verify the sequence-specific endonuclease activity of the assembled Cas9 RNP complex.
Research Reagent Solutions:
- Pre-assembled RNP Complex: From Protocol 1, step 2.
- Target DNA Substrate: A 200-500 bp PCR-amplified dsDNA fragment containing the target sequence and appropriate PAM. Purify via spin column.
- 10X Cleavage Reaction Buffer: 500 mM NaCl, 100 mM Tris-HCl, 50 mM MgCl2, pH 7.9 at 25°C.
- Stop Solution: 50 mM EDTA, 50% Glycerol, 1% SDS, 0.1% Bromophenol Blue.
- Agarose Gel: 2% agarose in 1X TAE buffer with added SYBR Safe DNA stain.
- DNA Ladder: 100 bp ladder suitable for resolving cleavage fragments.
Procedure:
- Cleavage Reaction Setup: In a PCR tube, combine:
- 20 nM pre-assembled RNP (from Protocol 1, step 2)
- 40 nM target DNA substrate
- 2 µL 10X Cleavage Reaction Buffer
- Nuclease-free water to 20 µL final volume.
- Mix gently and incubate in a thermal cycler or heat block at 37°C for 60 minutes.
- Reaction Termination: Add 5 µL of Stop Solution to each reaction. Incubate at 65°C for 10 minutes to inactivate Cas9 and denature proteins.
- Analysis by Gel Electrophoresis: Load 15-20 µL of the terminated reaction onto a pre-cast 2% agarose gel. Include appropriate controls (DNA substrate only, RNP only). Run at 120V for 45 minutes in 1X TAE buffer.
- Visualization: Image the gel using a standard gel doc system. A successful cleavage is indicated by the disappearance of the full-length substrate band and the appearance of two smaller, discrete bands corresponding to the predicted cleavage products.
Diagrams
Diagram Title: Workflow for Validating RNP Pre-Delivery
Diagram Title: Mechanism of Cas9 RNP Cleavage
The Scientist's Toolkit
Table 3: Essential Reagents for RNP QC Assays
| Item | Function | Critical for Assay |
|---|---|---|
| Recombinant Cas9 Nuclease (Purified) | Catalytic protein component of the RNP. Must be nuclease-free and in a known storage buffer. | Gel Shift, In Vitro Cleavage |
| Chemically Modified sgRNA | Provides target specificity and stability. Synthetic, HPLC-purified guides reduce batch variability. | Gel Shift, In Vitro Cleavage |
| 10X Cas9-sgRNA Formation Buffer | Optimized buffer (with KCl, Mg2+, DTT) to promote proper protein-RNA folding and complex stability. | Gel Shift |
| 10X Cleavage Reaction Buffer | Provides optimal ionic strength and Mg2+ concentration for Cas9's catalytic activity on dsDNA. | In Vitro Cleavage |
| SYBR Gold Nucleic Acid Stain | Highly sensitive, fluorescent dye for detecting RNA and dsDNA in gels. Safer alternative to ethidium bromide. | Gel Shift, In Vitro Cleavage |
| Pre-cast Native PAGE Gels (6%) | Ensure consistent pore size and buffer conditions for reproducible separation of protein, RNA, and RNP complexes. | Gel Shift |
| Target DNA Substrate (PCR-amplified) | Validates RNP function. Must contain an exact match to the gRNA spacer and an appropriate PAM sequence. | In Vitro Cleavage |
| Nuclease-free Water & Tubes | Prevents degradation of RNA and substrate DNA by environmental nucleases. | All Steps |
Within the critical thesis context of optimizing Cas9 protein-sgRNA ribonucleoprotein (RNP) complex formation for primary cell genome editing, robust post-editing analysis is paramount. Primary cells, with their limited expansion capacity and heterogeneity, demand efficient, multiplexed analytical techniques to accurately quantify editing outcomes and functional consequences. This application note details established and emerging best practices for three cornerstone analytical methods: Next-Generation Sequencing (NGS), T7 Endonuclease I (T7E1) assay, and Flow Cytometry.
Table 1: Comparative Analysis of Post-Editing Assessment Methods
| Parameter | Next-Generation Sequencing (NGS) | T7 Endonuclease I (T7E1) Assay | Flow Cytometry |
|---|---|---|---|
| Primary Readout | Nucleotide-level sequence variation; precise indel spectrum. | Detection of heteroduplex mismatches; estimates indel frequency. | Protein expression or marker presence at single-cell level. |
| Quantitative Data (Typical Sensitivity) | <0.1% variant allele frequency (VFE). | ~1-5% indel detection limit. | <0.1% for bright markers; depends on antibody. |
| Throughput | High (multiplexed samples). | Low to moderate (individual assays). | Very High (10^4-10^5 cells/sec). |
| Cost per Sample | High (decreasing with multiplexing). | Low. | Moderate (reagent-dependent). |
| Key Advantage | Unbiased, comprehensive sequence data. | Rapid, inexpensive, no specialized equipment. | Single-cell, multiparameter, phenotypic correlation. |
| Key Limitation | Cost, data complexity, turnaround time. | Low sensitivity, no sequence information, semi-quantitative. | Requires specific antibody or reporter; indirect for indels. |
| Best Application in Thesis Context | Definitive validation of editing efficiency & precise profiling of on-/off-target edits from RNP delivery. | Initial, rapid screening of RNP complex activity across multiple sgRNA designs. | Functional assessment of edits affecting surface/epitope expression (e.g., KO, KI). |
Detailed Experimental Protocols
Protocol 1: Targeted NGS for Editing Efficiency and Specificity
Objective: To precisely quantify Cas9-sgRNA RNP-induced indel frequencies and spectra at on-target and predicted off-target loci in primary cells.
Materials (Research Reagent Solutions):
- Genomic DNA Extraction Kit: (e.g., Qiagen DNeasy Blood & Tissue Kit). Function: High-quality, PCR-inhibitor-free gDNA isolation.
- PCR Primers: Designed to amplify ~300-500bp region flanking target site.
- High-Fidelity PCR Master Mix: (e.g., NEB Q5). Function: Minimizes PCR-induced errors for accurate variant calling.
- Library Preparation Kit: (e.g., Illumina Nextera XT). Function: Attaches sequencing adapters and sample barcodes.
- SPRIselect Beads: (e.g., Beckman Coulter). Function: Size selection and purification of DNA fragments.
- Bioinformatics Pipeline: CRISPResso2, Cas-Analyzer, or custom alignment/variant calling scripts.
Methodology:
- gDNA Isolation: Harvest edited primary cells 72-96 hours post-RNP delivery. Extract gDNA per kit protocol. Quantify via fluorometry.
- Primary PCR Amplification: Perform first-round PCR with target-specific primers containing partial adapter sequences. Use 50-100ng gDNA per reaction. Cycle conditions: 98°C/30s; (98°C/10s, 65°C/30s, 72°C/30s) x 25-30 cycles; 72°C/2min.
- Library Indexing PCR: Use a second, limited-cycle (8-12 cycles) PCR to add full Illumina adapters and dual-index barcodes.
- Library Purification & Quantification: Clean amplicons with SPRIselect beads. Quantify library concentration via qPCR (e.g., KAPA Library Quant Kit).
- Sequencing: Pool libraries equimolarly. Sequence on an Illumina MiSeq or iSeq with 2x250bp or 2x300bp paired-end runs to ensure coverage across cut site.
- Data Analysis: Demultiplex reads. Align to reference genome. Use CRISPResso2 to quantify indel percentages and visualize decomposition plots centered on the target site.
Protocol 2: T7 Endonuclease I (T7E1) Mismatch Cleavage Assay
Objective: To rapidly assess the presence of Cas9-induced indels in a population of edited primary cells.
Materials (Research Reagent Solutions):
- PCR Reagents: Standard Taq polymerase, dNTPs, target-specific primers.
- PCR Purification Kit: For cleaning amplified products.
- T7 Endonuclease I Enzyme: (e.g., NEB #M0302L). Function: Recognizes and cleaves mismatched heteroduplex DNA.
- DNA Gel Loading Dye & Agarose: For electrophoresis analysis.
- Gel Imaging System: For quantification of band intensities.
Methodology:
- Amplicon Generation: Isolate gDNA. Amplify target region (~500-800bp) using standard PCR. Purify PCR product.
- Heteroduplex Formation: Dilute purified amplicon. Denature and reanneal using a thermal cycler program: 95°C/5min, ramp down to 85°C at -2°C/s, then ramp to 25°C at -0.1°C/s. This allows mismatches from wild-type/mutant strands to form.
- T7E1 Digestion: Mix reannealed DNA with NEBuffer 2.1 and 1μL T7E1 enzyme. Incubate at 37°C for 15-60 minutes.
- Analysis by Electrophoresis: Run digested products on a 2-2.5% agarose gel. Include undigested control.
- Quantification: Image gel. Use ImageJ or similar to quantify band intensities. Calculate indel frequency using formula: % Indel = 100 x (1 - sqrt(1 - (b+c)/(a+b+c))), where
ais integrated intensity of undigested band, andb&care cleavage products.
Protocol 3: Flow Cytometric Analysis of Editing Outcomes
Objective: To measure the functional consequence of editing (e.g., knockout, knock-in) in single, live primary cells.
Materials (Research Reagent Solutions):
- Fluorophore-conjugated Antibodies: Specific to the target cell surface protein for knockout validation.
- Viability Dye: (e.g., propidium iodide, DAPI, LIVE/DEAD fixable dyes). Function: Exclude dead cells from analysis.
- Cell Staining Buffer: PBS with 2-5% FBS.
- Fixation/Permeabilization Buffer Kit: (e.g., BD Cytofix/Cytoperm) if intracellular staining is required.
- Flow Cytometer: Capable of detecting relevant fluorophores.
Methodology:
- Cell Harvest & Preparation: Harvest edited primary cells 5-7 days post-editing to allow for protein turnover. Create single-cell suspension. Filter through a 40μm strainer.
- Viability Staining: Resuspend cell pellet in staining buffer containing viability dye. Incubate 10-20 min on ice, protected from light.
- Surface Antigen Staining: Wash cells. Resuspend in staining buffer with titrated antibody cocktail. Incubate 30 min on ice, protected from light.
- Wash & Fix: Wash cells twice. If needed, fix cells with 1-4% paraformaldehyde for 15 min on ice. For intracellular targets, perform permeabilization and subsequent staining steps per kit protocol.
- Acquisition & Analysis: Resuspend in staining buffer. Acquire data on flow cytometer, collecting >10,000 live, single-cell events. Use FSC-A vs FSC-H to gate singlets. Use fluorescence-minus-one (FMO) controls to set gates for positive/negative populations. Calculate percentage of edited (e.g., knockout) cells.
Visualizations
Diagram 1: Post-Editing Analysis Decision Workflow
Diagram 2: Cas9 RNP Editing & Analysis Pathway
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Post-Editing Analysis of Primary Cells
| Reagent / Solution | Category | Critical Function in Analysis |
|---|---|---|
| Cas9 Nuclease, S. pyogenes (WT) | Core Enzyme | Forms active RNP complex with sgRNA for targeted DSB generation. |
| Synthetic sgRNA (chemically modified) | Targeting Guide | Confers DNA target specificity to Cas9; modified for stability in primary cells. |
| Electroporation/Transfection Reagent | Delivery | Enables efficient, low-toxicity RNP delivery into hard-to-transfect primary cells. |
| High-Fidelity PCR Polymerase (Q5, KAPA HiFi) | NGS Prep | Ensures accurate amplification of target loci for sequencing, minimizing PCR errors. |
| Illumina-Compatible Library Prep Kit | NGS Prep | Prepares amplicons for sequencing by adding adapters and sample-specific barcodes. |
| T7 Endonuclease I | Cleavage Assay | Binds and cleaves mismatched heteroduplex DNA, enabling detection of indels. |
| Fluorophore-Conjugated Validation Antibody | Flow Cytometry | Binds specifically to target protein for detection of knockout/knock-in by flow. |
| Viability Stain (Fixable LIVE/DEAD) | Flow Cytometry | Distinguishes live from dead cells during analysis, ensuring data quality. |
| Genomic DNA Extraction Kit | Sample Prep | Provides pure, high-molecular-weight gDNA from limited primary cell samples. |
This application note, framed within a thesis on Cas9-sgRNA ribonucleoprotein (RNP) complex formation, compares three genome-editing modalities for primary cells. Primary cells, with their limited expansion capacity and sensitivity, pose unique challenges for achieving high-efficiency, low-toxicity editing. The table below summarizes key performance metrics from recent literature.
Table 1: Comparative Performance of Editing Platforms in Diverse Primary Cell Types
| Platform | Typical Editing Efficiency (Range) | Indel/Frameshift Frequency | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Cas9-sgRNA RNP (NHEJ) | 20-80% (cell type dependent) | High (Primary product) | Rapid delivery, transient exposure, low off-target vs. plasmid, high knock-out efficiency. | Predominantly generates indels; limited precise editing. |
| Cas9-sgRNA RNP (HDR) | 0.5-20% (usually <5%) | Often accompanies HDR event | Can mediate precise knock-in with donor template. | Very low efficiency in most primary cells; requires active cycling. |
| Base Editor RNP (CBE/ABE) | 10-60% (at target base) | Very Low (<1% typically) | High-efficiency, precise single-base changes without DSBs or donor templates; works in non-dividing cells. | Restricted to specific transition mutations (C•G to T•A or A•T to G•C); potential bystander editing. |
| Prime Editor RNP (PE) | 1-30% (highly variable) | Low | Versatile; can mediate all 12 base-to-base conversions, small insertions, deletions; no DSBs. | Lower efficiency in primary cells; large RNP complex; complex pegRNA design. |
Table 2: Practical Delivery & Workflow Considerations for Primary Cells
| Parameter | Cas9-sgRNA RNP | Base Editor RNP | Prime Editor RNP |
|---|---|---|---|
| Common Delivery Method | Electroporation (e.g., Neon, 4D-Nucleofector) | Electroporation | Electroporation |
| Typical Complex Components | Cas9 protein + sgRNA | BE protein (e.g., BE4max) + sgRNA | PE protein (e.g., PEmax) + pegRNA + optional nicking sgRNA |
| Time to Genotype Analysis | Fast (48-72h post-delivery) | Fast (48-72h post-delivery) | May require longer (5-7 days) for turnover |
| Primary Readout | T7E1/SURVEYOR, NGS for indels | NGS for base conversion, targeted RFLP | NGS for targeted sequence changes |
Experimental Protocols
Protocol A: RNP Assembly & Electroporation for Primary Human T Cells (Cas9/BE/PE)
This core protocol is adaptable for all three platforms by substituting the specific protein and guide RNA components.
I. Materials: The Scientist's Toolkit
| Research Reagent Solution | Function/Explanation |
|---|---|
| Recombinant HiFi Cas9, BE4max, or PEmax Protein | High-specificity, high-activity, endotoxin-free protein for RNP formation. |
| Chemically Modified sgRNA/pegRNA | Synthetic guide RNA with 2'-O-methyl 3' phosphorothioate modifications enhances stability and reduces immune response. |
| Primary Cell Electroporation Buffer (e.g., P3) | Cell-type specific, low-cytotoxicity buffer for nucleofection. |
| Nucleofector Device (e.g., 4D-Nucleofector X Unit) | Enables high-efficiency RNP delivery into sensitive primary cells. |
| IL-2 (Interleukin-2) | Cytokine for T cell activation and expansion post-electroporation. |
| Genomic DNA Extraction Kit (Magnetic Bead-based) | For rapid, high-quality DNA extraction from limited cell numbers. |
| NGS Library Prep Kit for Amplicon Sequencing | For quantitative, unbiased assessment of editing outcomes and purity. |
II. Procedure
- Primary Human T Cell Isolation & Activation: Isolate CD3+ T cells from PBMCs using a negative selection kit. Activate with CD3/CD28 Dynabeads for 48 hours in RPMI-1640 with 10% FBS and 100 U/mL IL-2.
- RNP Complex Assembly:
- For Cas9/BE: Combine 10 µg (≈60 pmol) of protein with 6 µg (≈60 pmol) of modified sgRNA in duplex buffer. Incubate at 25°C for 10 minutes.
- For PE: Combine 15 µg (≈60 pmol) of PEmax protein with 9 µg (≈60 pmol) of modified pegRNA and 3 µg (≈30 pmol) of nicking sgRNA (if using). Incubate at 25°C for 10 minutes.
- Electroporation:
- Harvest 1e5 to 1e6 activated T cells per condition.
- Resuspend cell pellet in 20 µL of pre-warmed P3 buffer.
- Mix cells with the prepared RNP complex. Transfer to a 20 µL nucleofection cuvette.
- Run the appropriate pulse code (e.g., EH-115 for human T cells).
- Immediately add 80 µL of pre-warmed, cytokine-supplemented media to the cuvette and transfer cells to a culture plate.
- Post-Electroporation Culture: Culture cells in complete medium with IL-2 (200 U/mL). Assess viability at 24h. Expand cells as needed.
- Genotyping (72h-7d post-editing):
- Extract genomic DNA.
- Amplify the target locus by PCR using high-fidelity polymerase.
- Purify PCR amplicons and subject to next-generation amplicon sequencing.
- Analyze sequences using tools like CRISPResso2 (for Cas9), BE-Analyzer (for BEs), or prime-editing-analyzer (for PEs) to quantify editing efficiencies, indels, and product purity.
Protocol B: HDR Donor Co-delivery for Cas9 RNP in Primary Hematopoietic Stem/Progenitor Cells (HSPCs)
This protocol outlines the key modifications for knock-in experiments.
- RNP Assembly: Follow Protocol A step 2 for Cas9 RNP assembly.
- Donor Template Preparation: Use a single-stranded oligodeoxynucleotide (ssODN) or an AAV6 vector as the donor template. For ssODN, add at a final concentration of 1-5 µM to the RNP-cell mixture prior to electroporation. For AAV6, transduce cells 4-24 hours after electroporation.
- Electroporation of HSPCs: Use 1e5 CD34+ cells per condition and the DZ-100 pulse code with P3 buffer. Include a "RNP-only" control.
- Culture & Analysis: Culture cells in StemSpan with cytokines. Harvest cells at day 3-5 for initial NGS assessment of HDR and at later timepoints (e.g., day 14) for progenitor-derived colony assays to assess editing in clonogenic potential.
Visualized Workflows & Pathway Logic
Title: Platform Selection Logic for Primary Cell Editing
Title: Stepwise RNP Editing Protocol for Primary Cells
Title: Molecular Mechanisms of Cas9, BE, and PE Platforms
Application Notes
In primary cell editing research, the formation of a functional Cas9-sgRNA ribonucleoprotein (RNP) complex is the critical first step that determines all downstream editing outcomes. Quantitative benchmarking of these outcomes—specifically, Insertion/Deletion (Indel) percentage and the rate of Biallelic Knockout (KO)—is non-negotiable for evaluating RNP efficacy, guiding sgRNA design, and translating findings into therapeutic applications. These metrics directly reflect the efficiency and completeness of gene disruption, informing decisions on experimental scalability and preclinical development.
Accurate measurement requires a combination of endpoint and next-generation sequencing (NGS) analyses. While T7 Endonuclease I (T7E1) or Surveyor assays offer rapid, cost-effective estimates of Indel %, they lack sensitivity for low-efficiency edits and cannot delineate complex allele distributions. For definitive quantification, especially in heterogeneous primary cell populations, NGS of the target locus is the gold standard. It provides single-nucleotide resolution, enabling the precise calculation of Indel frequency and the critical distinction between monoallelic and biallelic modifications. The following protocols and data frameworks are designed to standardize this quantification within the context of Cas9-sgRNA complex optimization.
Table 1: Comparison of Methods for Quantifying Editing Outcomes
| Method | Primary Readout | Sensitivity | Throughput | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| T7E1 / Surveyor Assay | Estimated Indel % via gel electrophoresis | Low (~1-5%) | Low | Fast, inexpensive, no specialized equipment | Semi-quantitative, cannot detect precise edits or biallelic status |
| Sanger Sequencing + Decomposition | Indel % via trace deconvolution | Moderate (~1%) | Low-Moderate | Low-cost sequencing, provides sequence context | Accuracy drops with polyclonality >~30%; biallelic inference is indirect |
| Next-Generation Sequencing (NGS) | Precise Indel %, Biallelic KO % | Very High (<0.1%) | High | Nucleotide-resolution, quantitative, direct biallelic analysis | Higher cost, complex data analysis, longer turnaround |
Table 2: Example NGS Data Output from Primary T-cell Editing (Target: PDCD1)
| Sample | sgRNA | RNP Dose (pmol) | Total Reads | Indel % | Biallelic KO % | Monoallelic Edit % | Wild-Type % |
|---|---|---|---|---|---|---|---|
| Donor 1, Rep 1 | sgPD-1_A | 10 | 85,422 | 78.5% | 65.2% | 13.3% | 21.5% |
| Donor 1, Rep 2 | sgPD-1_A | 10 | 79,105 | 76.8% | 63.7% | 13.1% | 23.2% |
| Donor 1, Control | NTC | 0 | 81,997 | 0.2% | 0.0% | 0.2% | 99.8% |
| Donor 2, Rep 1 | sgPD-1_B | 10 | 88,111 | 45.3% | 28.9% | 16.4% | 54.7% |
Experimental Protocols
Protocol 1: Formation and Delivery of Cas9-sgRNA RNP Complex for Primary Cells Objective: To form active RNPs and introduce them into primary cells (e.g., T cells, HSCs) via electroporation.
- Complex Formation: Dilute purified, high-quality Cas9 protein (e.g., SpyFi Cas9) and synthesized, chemically modified sgRNA to working concentrations in sterile, nuclease-free buffer. Combine at a molar ratio of 1:2.5 (Cas9:sgRNA). Incubate at room temperature for 10-20 minutes to allow RNP assembly.
- Cell Preparation: Isolate and activate primary cells as required. Wash cells thoroughly with electroporation buffer or PBS without Ca2+/Mg2+. Resuspend cells at a high concentration (e.g., 1-2 x 10^8 cells/mL).
- Electroporation: Mix the RNP complex with the cell suspension. Transfer to an appropriate electroporation cuvette or strip. Electroporate using a primary cell-optimized program (e.g., on a Lonza 4D-Nucleofector or Bio-Rad Gene Pulser).
- Recovery: Immediately add pre-warmed culture medium and transfer cells to a culture plate. Incubate at 37°C, 5% CO2. Allow 48-72 hours for editing outcomes to manifest before genomic DNA (gDNA) harvest.
Protocol 2: Genomic DNA Harvest and Amplicon Library Preparation for NGS Objective: To isolate gDNA and generate sequencing libraries from the edited target locus.
- gDNA Extraction: At 72 hours post-editing, pellet ~1x10^5 to 1x10^6 cells. Extract gDNA using a silica-column or magnetic bead-based kit. Quantify gDNA by fluorometry.
- Primary PCR (Amplification of Target Locus): Design primers ~150-250bp flanking the Cas9 cut site. Perform PCR using a high-fidelity polymerase. Include sample-specific barcodes (if not adding in a second step).
- Reaction Setup: 50-100ng gDNA, 0.5µM primers, 1x HF buffer, 200µM dNTPs, 1U polymerase. Cycle: 98°C 30s; 30x (98°C 10s, 60°C 20s, 72°C 20s); 72°C 2 min.
- Library Indexing and Purification: Clean up the primary PCR product with magnetic beads. Perform a second, limited-cycle PCR to add full Illumina adapter sequences (P5/P7) and unique dual indices (UDIs). Purify the final library with bead-based size selection (e.g., 0.8x ratio).
- Sequencing: Quantify the library by qPCR. Pool libraries at equimolar ratios and sequence on an Illumina MiSeq or iSeq system using a 2x150bp or 2x250bp paired-end run to ensure coverage across the cut site.
Protocol 3: Bioinformatic Analysis for Indel % and Biallelic KO Calculation Objective: To process NGS data and calculate key editing metrics.
- Demultiplexing & Quality Control: Use
bcl2fastqorbcl-convertto generate FASTQ files. Assess read quality with FastQC. - Read Alignment & Processing: Align paired-end reads to the reference genome sequence (amplicon) using a sensitive aligner like
BWA-MEM. Process SAM/BAM files withsamtools(sort, index). Identify the cut site coordinate. - Indel Quantification: Use a specialized tool like
CRISPResso2orcas9-analyzer.- CRISPResso2 Command:
CRISPResso2 -r1 sample_R1.fastq.gz -r2 sample_R2.fastq.gz -a CCTCTACTCCACCTATC... -g GATCCAGTTCGAGTCAG --quantification_window_size 20 -w 10 - The tool aligns reads, centers them on the predicted cut site, and reports the percentage of reads containing insertions or deletions within the quantification window.
- CRISPResso2 Command:
- Biallelic KO Calculation: Analyze the allele table output from CRISPResso2. An allele is considered "modified" if it contains an Indel within the coding sequence that causes a frameshift. Biallelic KO % is calculated as the percentage of reads where all aligned reads from a single cell (inferred statistically) contain frameshift mutations on both alleles. This is often approximated from the NGS data as: % Biallelic KO = (1 - (sqrt(Fraction of Unmodified Reads))) * (Total Edited Reads Fraction), or more directly, by identifying reads with two distinct frameshift alleles or a homozygous frameshift mutation.
Visualization
Diagram Title: Workflow for RNP Editing and Outcome Quantification
Diagram Title: Genotype Outcomes Leading to Biallelic Knockout
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for RNP Editing & Benchmarking
| Item | Function & Importance |
|---|---|
| High-Purity Cas9 Nuclease | Endonuclease that, when complexed with sgRNA, creates a DSB. Essential for efficient cleavage with minimal off-target effects. |
| Chemically Modified sgRNA (e.g., 2'-O-Methyl, Phosphorothioate) | Increases RNP stability, reduces innate immune response in primary cells, and improves editing efficiency. |
| Primary Cell Electroporation Kit | Optimized buffers and protocols (e.g., Lonza P3/P5, Thermo Neon) for delivering RNP into sensitive primary cells with high viability. |
| High-Fidelity PCR Master Mix | For accurate, low-error amplification of the target locus from limited gDNA, critical for clean NGS library prep. |
| Illumina-Compatible Dual-Indexed UMI Adapters | Enables multiplexing of samples and identification of PCR duplicates, improving quantification accuracy. |
| CRISPResso2 Software | Standardized, comprehensive bioinformatics pipeline for quantifying Indel frequencies and editing outcomes from NGS data. |
| NGS Purification Beads (SPRI) | For size selection and clean-up during amplicon library preparation, removing primer dimers and non-specific products. |
Thesis Context: This document details application notes and protocols for the long-term validation of primary cells edited via Cas9 protein:sgRNA ribonucleoprotein (RNP) complexes. The methods are framed within the broader thesis that the kinetics, stability, and delivery method of pre-formed Cas9 RNP complexes are critical for achieving high-fidelity edits while minimizing off-target effects and preserving primary cell viability and function over extended culture periods.
Table 1: Key Quantitative Metrics for Long-term Validation of Edited Primary Cells
| Validation Category | Specific Assay | Typical Time Points Post-Editing | Key Quantitative Readout | Acceptance Benchmark | |
|---|---|---|---|---|---|
| Genomic Stability | Off-target Analysis (GUIDE-seq) | 2-3 days, 4 weeks | Number of significant off-target sites identified | ≤ 2 sites with ≥ 0.1% INDEL frequency | |
| Karyotyping / CNV Analysis | 2 weeks, 8 weeks | Chromosomal aberrations; Copy Number Variations | No novel aberrations vs. unedited control | ||
| TP53 Activation (p21 mRNA) | 3 days, 1 week | Fold-change in p21 expression (qPCR) | < 2-fold increase vs. control | ||
| Editing Persistence | Target Site INDEL Efficiency (NGS) | 3 days (initial), 4, 8, 12 weeks | % INDELs at on-target locus | Stability within ±15% of initial efficiency | |
| Functional Phenotype | Cell-type Specific Function Assay (e.g., Cytokine Secretion) | Weekly for up to 12 weeks | Concentration (e.g., pg/mL IFN-γ) | No significant decline vs. unedited functional cells | |
| Proliferation / Senescence | Weekly population doublings | Cumulative Population Doublings; β-galactosidase+ % | Rate not significantly lower than control | ||
| Single-cell Clonal Analysis | At 4 weeks post-editing | Clonal diversity; phenotype-genotype linkage | > 80% of clones maintain desired edit & function |
Detailed Protocols
Protocol 1: Longitudinal Tracking of On-target Editing & Clonal Composition in Primary T-cells
Objective: To monitor the stability of editing outcomes and clonal dynamics over 12+ weeks of culture. Materials: Edited primary human T-cells, IL-2, X-VIVO 15 media, activated CD3/CD28 beads, genomic DNA extraction kit, on-target NGS amplicon assay.
Procedure:
- Cell Culture & Expansion: Post-electroporation with Cas9 RNP, expand T-cells in IL-2 (100 IU/mL) supplemented media with periodic reactivation using CD3/CD28 beads every 14 days.
- Sampling: At T=3 days, 4, 8, and 12 weeks, extract genomic DNA from a minimum of 1e5 cells using a column-based kit.
- NGS Library Prep:
- Amplify the target locus using barcoded primers (15-30ng gDNA per reaction).
- Purify PCR products and quantify.
- Pool equimolar amounts of amplicons for sequencing on an Illumina MiSeq (2x300 bp).
- Data Analysis: Use CRISPResso2 to quantify INDEL frequencies and distributions. Track changes in the top 10 INDEL sequences over time to assess clonal drift.
Protocol 2: Assessment of Genomic Stability via nuclease-independent CNV Analysis
Objective: To identify large-scale genomic aberrations induced by editing or clonal expansion. Materials: KaryoStat+ or similar CMA assay, genomic DNA from edited and control cells at week 8.
Procedure:
- DNA Preparation: Isolate high-quality genomic DNA (≥ 250ng, concentration ≥ 20ng/μL, A260/280 ~1.8).
- Labeling & Hybridization: Fragment DNA, label with biotin, and hybridize to the CNV array according to manufacturer’s instructions.
- Washing & Scanning: Wash arrays stringently and scan using a compatible microarray scanner.
- Analysis: Use provided software (e.g., Chromosome Analysis Suite) to call CNVs. Compare edited sample profiles to unedited isogenic controls to identify novel, large-scale (>50 kb) losses or gains.
Protocol 3: Functional Phenotype Assay – Antigen-Specific CD8+ T-cell Activation
Objective: To validate that edited primary T-cells retain their antigen-specific cytotoxic function. Materials: Edited TCR- or CAR-expressing CD8+ T-cells, matched antigen-presenting cells, peptide antigen, flow cytometry antibodies (CD107a, IFN-γ, TNF-α), GolgiPlug.
Procedure:
- Co-culture: Co-culture 1e5 edited T-cells with antigen-pulsed APCs (1:1 ratio) for 6 hours in the presence of CD107a antibody and GolgiPlug.
- Staining & Fixation: Harvest cells, stain surface markers, then fix/permeabilize.
- Intracellular Staining: Stain for IFN-γ and TNF-α.
- Flow Cytometry: Acquire data on a flow cytometer. Analyze the percentage of CD8+ cells positive for CD107a and/or cytokines. Compare to unedited T-cells with the same specificity at multiple time points (e.g., weeks 2, 6, 10).
Visualizations
Diagram 1: Workflow for Long-term Validation of Edited Primary Cells
Diagram 2: Key Signaling Pathways Monitoring Genomic Stress in Edited Cells
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Supplier Examples | Function in Long-term Validation |
|---|---|---|
| Cas9 Nuclease (WT), HPLC-purified | IDT, Thermo Fisher, Synthego | High-quality protein for RNP formation ensures high on-target activity and low toxicity in sensitive primary cells. |
| Chemically Modified sgRNA (ALT-R) | IDT | Enhances RNP stability and reduces immunogenicity, critical for functional assays in immune cells. |
| Primary Cell Electroporation Kit | Lonza (P3 kit), Bio-Rad (Gene Pulser) | Enables high-efficiency, low-toxicity delivery of Cas9 RNPs into hard-to-transfect primary cells (T-cells, HSCs). |
| GUIDE-seq Kit | Integrated DNA Technologies | Unbiased detection of off-target cleavage sites early post-editing to inform genomic stability risk. |
| Cell Culture Media (X-VIVO, StemSpan) | Lonza, STEMCELL Technologies | Serum-free, optimized media supports long-term expansion and maintenance of primary cell phenotypes. |
| Cytokine/Growth Factor Cocktails | PeproTech, R&D Systems | Essential for maintaining viability, promoting expansion, and preserving lineage-specific functions over time. |
| NGS Amplicon-EZ Service | Genewiz, Azenta | Streamlined deep sequencing of target loci for quantitative, longitudinal tracking of INDEL frequencies. |
| Copy Number Variation Array | Affymetrix (KaryoStat) | Provides genome-wide, nuclease-independent assessment of large-scale genomic integrity. |
| Flow Cytometry Antibody Panels | BioLegend, BD Biosciences | Multiplexed detection of surface markers, intracellular cytokines, and functional markers for phenotyping. |
Conclusion
Effective Cas9-sgRNA complex formation and delivery represent the cornerstone of successful genome editing in primary cells. By mastering the foundational biochemistry, adopting robust and optimized assembly protocols, systematically troubleshooting inefficiencies, and implementing rigorous validation, researchers can overcome the inherent challenges of these delicate systems. As the field advances, the refinement of RNP-based editing—coupled with emerging technologies like high-fidelity Cas variants and novel delivery vehicles—promises to unlock the full therapeutic potential of primary cell engineering, accelerating the development of next-generation cell therapies and ex vivo gene corrections for clinical applications.