Precision Control in Immunology: Mastering CRISPRa Activation and CRISPRi Interference in Primary Immune Cells
This article provides a comprehensive guide for researchers and drug developers on the application of CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) technologies in primary immune cells.
Precision Control in Immunology: Mastering CRISPRa Activation and CRISPRi Interference in Primary Immune Cells
Abstract
This article provides a comprehensive guide for researchers and drug developers on the application of CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) technologies in primary immune cells. It covers foundational principles, detailing the mechanisms of dCas9-VPR, dCas9-KRAB, and newer engineered systems. A deep dive into optimized protocols for delivery, screening, and phenotypic analysis in T cells, macrophages, and other primary immune subsets is presented. Critical troubleshooting advice addresses common challenges like low efficiency, cytotoxicity, and off-target effects. Finally, the article compares CRISPRa/i to other perturbation methods and outlines validation strategies, highlighting transformative applications in immunology research, therapeutic target discovery, and the engineering of next-generation cell therapies.
CRISPRa and CRISPRi Fundamentals: Redefining Immune Cell Perturbation
Application Notes
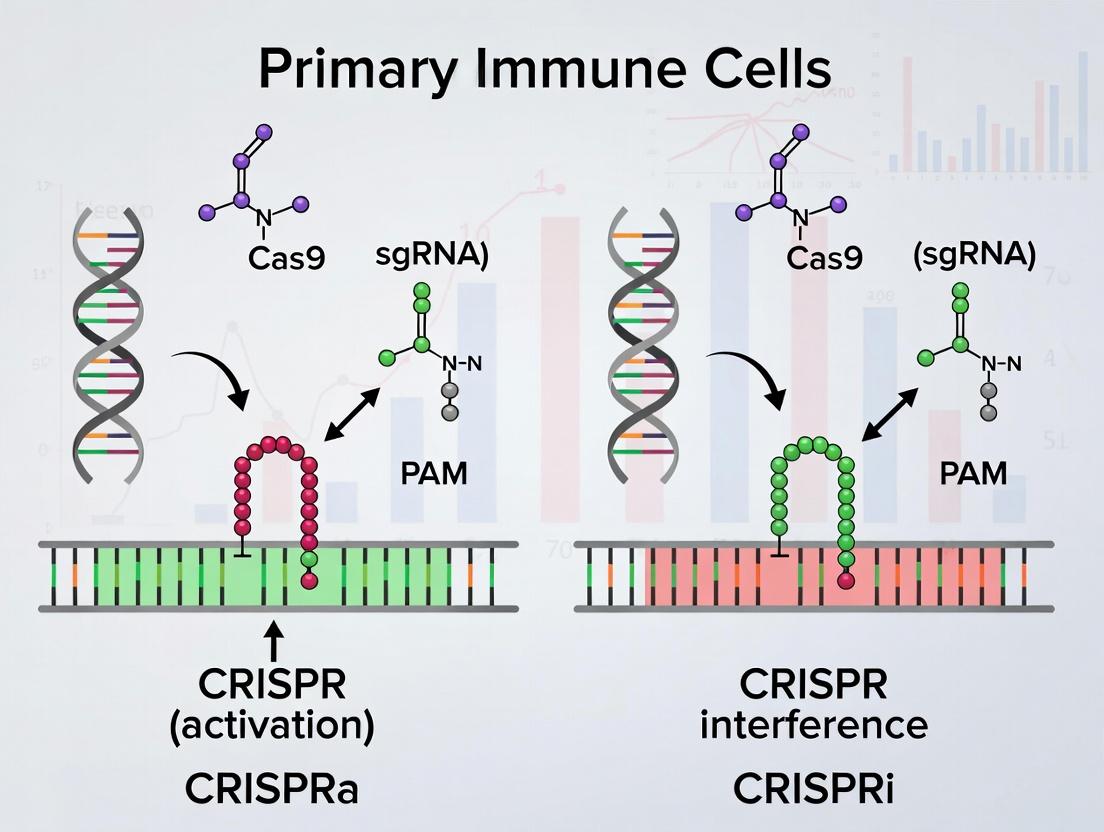
CRISPR activation (CRISPRa) and interference (CRISPRi) have revolutionized functional genomics in hard-to-transfect primary immune cells, such as T cells, B cells, and macrophages. These systems, built upon a catalytically dead Cas9 (dCas9), enable precise, programmable transcriptional modulation without altering the underlying DNA sequence. This is critical for dissecting gene regulatory networks in immunity, validating therapeutic targets, and engineering cell states for immunotherapy.
dCas9 as a Modular Scaffold: The core component is dCas9, a nuclease-deficient version of Streptococcus pyogenes Cas9 (D10A and H840A mutations). It retains its ability to bind specific DNA sequences guided by a single guide RNA (sgRNA) but does not create double-strand breaks. This makes it a versatile platform for recruiting effector domains to genomic loci.
CRISPRa with Synergistic Activators: To achieve robust transcriptional activation, multiple, synergistic activator domains are fused to dCas9. Two primary systems are widely used:
- VPR: A tripartite activator comprising VP64, p65, and Rta. VP64 recruits general transcription factors, while p65 and Rta provide co-activator functions, leading to strong, synergistic gene upregulation.
- SAM (Synergistic Activation Mediator): A more complex system where dCas9 is fused only to VP64. The sgRNA is modified with two MS2 RNA aptamers, which recruit MCP-p65-HSF1 fusion proteins. This creates a highly potent, multi-component activation complex.
CRISPRi with Repressive Effectors: For gene repression, repressor domains are fused to dCas9. The most common is the KRAB (Krüppel-associated box) domain from KOX1. When recruited to a promoter or enhancer, KRAB mediates the establishment of heterochromatin, leading to durable and specific transcriptional silencing.
Quantitative Performance in Primary Immune Cells: Recent studies have optimized delivery (lentiviral/AAV vectors, electroporation of RNP) and expression parameters for primary cells. Key performance metrics are summarized below.
Table 1: Quantitative Performance of CRISPRa/i Systems in Primary Immune Cells
| System | Effector Domain | Typical Fold Change (mRNA) | Optimal Targeting Region | Key Delivery Method | Notable Immune Cell Application |
|---|---|---|---|---|---|
| CRISPRa (VPR) | dCas9-VP64-p65-Rta | 10x - 500x | -200 to -50 bp from TSS | Lentivirus, mRNA Electroporation | Activating cytokine genes (IL-2, IFN-γ) in primary T cells |
| CRISPRa (SAM) | dCas9-VP64 + MS2-p65-HSF1 | 100x - 2000x | -200 to +1 bp from TSS | Lentivirus | Pooled activation screens for surface receptor identification (e.g., CD markers) |
| CRISPRi (KRAB) | dCas9-KRAB | 5x - 100x (repression) | -50 to +300 bp from TSS | Lentivirus, RNP Electroporation | Silencing checkpoint inhibitors (PD-1, CTLA-4) in exhausted T cells |
Experimental Protocols
Protocol 1: Lentiviral Delivery of dCas9-VPR for Gene Activation in Primary Human T Cells
Objective: To achieve stable, inducible activation of an endogenous immunomodulatory gene. Materials: See "The Scientist's Toolkit" below. Procedure:
- sgRNA Design & Cloning: Design two sgRNAs targeting the promoter region (-150 bp from TSS) of your gene of interest (GOI). Clone them into a lentiviral sgRNA expression vector (e.g., pLKO5.sgRNA-EFS-GFP).
- Lentivirus Production: Co-transfect HEK293T cells with your sgRNA vector, the dCas9-VPR expression vector (pHR-dCas9-VPR), and packaging plasmids (psPAX2, pMD2.G) using PEI transfection reagent.
- Virus Harvest & Concentration: Collect supernatant at 48h and 72h post-transfection. Concentrate using Lenti-X Concentrator.
- T Cell Isolation & Activation: Isolate CD3+ T cells from PBMCs using magnetic beads. Activate with CD3/CD28 Dynabeads (1:1 bead:cell ratio) in RPMI-1640 + 10% FBS + 100 U/mL IL-2.
- Transduction: 24h post-activation, transduce T cells with concentrated lentivirus in the presence of 8 µg/mL polybrene. Centrifuge at 800 x g for 90 min (spinoculation).
- Analysis: 72-96h post-transduction, assess activation by flow cytometry (if targeting a surface protein) or harvest RNA for qRT-PCR analysis of GOI mRNA levels. Normalize to a housekeeping gene (e.g., GAPDH) and compare to non-targeting sgRNA control.
Protocol 2: CRISPRi via RNP Electroporation for Rapid Gene Knockdown in Primary Macrophages
Objective: To achieve rapid, transient repression of a inflammatory gene in monocyte-derived macrophages (MDMs). Materials: See "The Scientist's Toolkit" below. Procedure:
- sgRNA Synthesis: Order chemically modified sgRNAs (2'-O-methyl 3' phosphorothioate) targeting the TSS (+50 bp) of your GOI. Resuspend in nuclease-free buffer.
- RNP Complex Formation: Combine 60 pmol of purified dCas9-KRAB protein with 120 pmol of sgRNA (1:2 molar ratio) in electroporation buffer. Incubate at room temperature for 10 min.
- Macrophage Differentiation: Isolate CD14+ monocytes from PBMCs. Differentiate in RPMI-1640 + 10% FBS + 50 ng/mL M-CSF for 5-7 days.
- Electroporation: Harvest MDMs, wash, and resuspend in P3 Primary Cell Buffer. Mix 2e5 cells with the pre-formed RNP complex. Electroporate using the 4D-Nucleofector (program DZ-167). Immediately add pre-warmed culture medium.
- Stimulation & Analysis: 24h post-electroporation, stimulate cells with LPS (100 ng/mL) for 6h. Harvest RNA and perform qRT-PCR to quantify repression of the GOI. Compare to cells electroporated with a non-targeting sgRNA RNP.
Visualization
Title: SAM CRISPRa Complex Assembly & Transcription Activation
Title: Mechanism Comparison: CRISPRi Silencing vs CRISPRa Activation
The Scientist's Toolkit
Table 2: Essential Research Reagents for CRISPRa/i in Primary Immune Cells
| Reagent / Material | Supplier Examples | Function in Experiment |
|---|---|---|
| dCas9-VPR Lentiviral Vector | Addgene (#63798), Sigma-Aldrich | Stable delivery of the core CRISPRa activator fusion protein. |
| dCas9-KRAB Protein (Nuclease-Free) | Aldevron, Thermo Fisher Scientific | Ready-to-use protein for forming RNP complexes for CRISPRi electroporation. |
| Chemically Modified sgRNA (synthego) | Synthego, IDT | Enhanced stability and reduced immunogenicity for RNP experiments in sensitive primary cells. |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene (#12260, #12259) | Essential for producing replication-incompetent lentiviral particles. |
| Lenti-X Concentrator | Takara Bio | Polyethylene glycol-based reagent for quick, simple lentivirus concentration. |
| Human CD3/CD28 Dynabeads | Thermo Fisher Scientific | For robust, reversible activation and expansion of primary human T cells. |
| Nucleofector 4D & P3 Kit | Lonza | Gold-standard electroporation system for high-efficiency RNP delivery into hard-to-transfect immune cells. |
| M-CSF (Human) | PeproTech | Cytokine required for the differentiation of monocytes into macrophages. |
| qPCR Assays for Immune Genes | Thermo Fisher Scientific (TaqMan), Bio-Rad | Validated primers/probes for accurate quantification of gene expression changes post-modulation. |
Why Primary Immune Cells? Unique Challenges and Opportunities Compared to Cell Lines.
Within the rapidly advancing field of immunology and immuno-oncology, the choice of cellular model is foundational. While immortalized cell lines have been the workhorse of basic research, primary immune cells—harvested directly from blood or tissue—offer unparalleled physiological relevance. This document, framed within a thesis on CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells, details the critical advantages, inherent challenges, and essential protocols for working with these dynamic systems compared to traditional cell lines.
Comparative Analysis: Primary Immune Cells vs. Immortalized Cell Lines
The decision to use primary cells or cell lines involves trade-offs between physiological fidelity and experimental convenience. The quantitative and qualitative differences are summarized below.
Table 1: Core Comparison of Primary Immune Cells and Immortalized Cell Lines
| Feature | Primary Immune Cells | Immortalized Cell Lines (e.g., Jurkat, THP-1) |
|---|---|---|
| Physiological Relevance | High; retain native receptor expression, signaling, heterogeneity, and functional responses. | Low; exhibit genetic and phenotypic drift, adapted to culture. |
| Genetic Stability | Normal diploid genome, but finite lifespan. | Often aneuploid; genetically unstable over long-term culture. |
| Heterogeneity | High; reflects donor variability and subset diversity (e.g., T cell subsets). | Low; clonal and homogeneous population. |
| Proliferation Capacity | Limited; most are non-dividing or require specific activation. | Unlimited; readily proliferate. |
| CRISPR Manipulation Efficiency | Typically low (5-40% for nucleofection); requires optimized protocols. | High (often >70-80%); easily transfected. |
| Cost & Accessibility | High cost; requires donor material, isolation kits, and fresh use. | Low cost; readily available from repositories. |
| Reproducibility | Subject to donor-to-donor variability. | High, within the same passage range. |
| Key Application | Translational research, functional studies, drug response testing, adoptive cell therapy. | High-throughput screening, mechanistic studies, protocol establishment. |
Table 2: Quantitative Metrics for CRISPR Delivery in Primary T Cells vs. Jurkat Cell Line
| Method | Primary Human T Cells (Efficiency % / Viability %) | Jurkat T Cell Line (Efficiency % / Viability %) | Notes |
|---|---|---|---|
| Electroporation (Nucleofection) | 40-75% / 50-70% | 80-95% / 70-85% | Gold standard for primary cells; requires specific kits. |
| Lentiviral Transduction | 30-60% (dividing cells) | >90% | Requires activation/proliferation for primary cells. |
| Lipofection | <5% / Variable | >80% / High | Generally ineffective for most primary immune cells. |
| mRNA RNP Delivery | 50-90% / 60-80% | N/A | CRISPR ribonucleoprotein complex; high efficiency, transient expression. |
Unique Challenges and Opportunities in Primary Immune Cell Research
The challenges of working with primary immune cells are significant but surmountable, and overcoming them unlocks unique opportunities, especially for CRISPR-based functional genomics.
Challenges:
- Low Transfection Efficiency: Traditional chemical methods are ineffective.
- Donor Variability: Requires robust experimental design with multiple donors.
- Cell Sensitivity: Prone to activation-induced cell death or anergy upon manipulation.
- Short Lifespan: Limits long-term assays, making stable CRISPRi/a delivery tricky.
- Complex Activation Requirements: Specific cytokines and co-stimulation are needed for function and proliferation.
Opportunities for CRISPRa/i:
- Define In Vivo Relevant Gene Function: Knockdown or overexpress genes in their native chromatin context.
- Functional Screens in Physiological Systems: Identify genes controlling immune cell trafficking, polarization, or tumor killing.
- Engineer Next-Generation Therapies: Use CRISPRa to enhance chimeric antigen receptor (CAR) T cell function or CRISPRi to silence immune checkpoints.
- Model Disease Variants: Introduce or correct patient-specific variants to study mechanisms.
Detailed Application Notes & Protocols
Protocol 1: Nucleofection of CRISPR RNP into Primary Human T Cells for CRISPRi/a
This protocol is optimized for transient delivery of a CRISPR ribonucleoprotein (RNP) complex, which minimizes off-target effects and toxicity, ideal for primary cells.
Research Reagent Solutions Toolkit:
| Item | Function/Description |
|---|---|
| Human T Cell Isolation Kit (e.g., Pan T Cell) | Negative selection to obtain untouched, resting T cells from PBMCs. |
| IL-2 (Recombinant Human) | Cytokine essential for T cell survival and proliferation post-activation. |
| Anti-CD3/CD28 Activator | Dynabeads or soluble antibody for stimulating T cell activation and division. |
| Nucleofector Device & Kit (e.g., P3 Kit) | Specialized electroporation system and buffer optimized for primary cells. |
| Alt-R S.p. HiFi Cas9 Nuclease | High-fidelity Cas9 for knockout, or dCas9-KRAB/VP64 for i/a. |
| Alt-R CRISPR-Cas9 tracrRNA & crRNA | Synthetic RNA components for RNP assembly; crRNA designed for target gene. |
| Electroporation Cuvettes | Disposable cuvettes for nucleofection. |
| Pre-warmed TexMACS or X-VIVO Media | Serum-free, low-cytokine media ideal for human T cell culture. |
Procedure:
- Isolate T Cells: Isolate primary T cells from healthy donor PBMCs using a negative selection isolation kit. Rest cells overnight in TexMACS medium with 5% human AB serum and 10 ng/mL IL-2.
- Activate T Cells: Stimulate cells with anti-CD3/CD28 activator (e.g., 1 bead per 2 cells) for 24-48 hours prior to nucleofection. This improves survival and editing efficiency.
- Assemble RNP Complex:
- Resuspend Alt-R crRNA and tracrRNA in nuclease-free buffer to 100 µM.
- Mix equal volumes (e.g., 1.5 µL each) of 100 µM crRNA and tracrRNA. Heat at 95°C for 5 min, then cool to room temperature to form the guide RNA (gRNA) duplex.
- For one reaction, mix 3 µL of 62 µM Alt-R Cas9 protein (or dCas9 protein) with 3 µL of the 50 µM gRNA duplex (final ratio 1:2.5 protein:gRNA). Incubate at room temperature for 15-20 min to form the RNP complex.
- Prepare Cells for Nucleofection: Harvest activated T cells, count, and centrifuge. Resuspend cell pellet in pre-warmed Nucleofector Solution from the kit to achieve 1-5 x 10^6 cells per 20 µL.
- Nucleofection: Combine 20 µL cell suspension with the pre-assembled 6 µL RNP complex. Transfer the entire volume into a nucleofection cuvette. Place cuvette in the Nucleofector device and run the recommended program (e.g., EH-115 for primary T cells). Immediately after pulse, add 80 µL of pre-warmed medium to the cuvette.
- Recovery and Culture: Gently transfer the cells from the cuvette to a pre-warmed culture plate containing complete TexMACS medium with IL-2 (50-100 U/mL). Place plate in a 37°C, 5% CO2 incubator. Assess viability and editing efficiency at 48-72 hours post-nucleofection via flow cytometry (for fluorescent reporter) or genomic analysis.
Protocol 2: Lentiviral Transduction for Stable dCas9 Expression in Primary T Cells
For prolonged CRISPRa/i studies, stable integration of the dCas9 effector is preferred.
Procedure:
- Produce Lentivirus: Generate 2nd or 3rd generation lentiviral particles encoding dCas9-KRAB (for CRISPRi) or dCas9-VP64 (for CRISPRa) and a selection marker (e.g., puromycin resistance or GFP) in 293T cells.
- Activate T Cells: Isolate and activate primary T cells with anti-CD3/CD28 as in Protocol 1, Step 2, for 24 hours.
- Transduction: On Retronectin-coated plates, add concentrated lentivirus at an appropriate MOI (Multiplicity of Infection; typically 5-20). Centrifuge activated T cells and resuspend in virus-containing medium with 8 µg/mL polybrene. Spin-infect by centrifuging the plate at 800 x g for 90 min at 32°C. Return to incubator.
- Selection and Expansion: 48 hours post-transduction, begin selection with puromycin (if applicable) or sort for GFP+ cells. Expand cells with IL-2 for 7-10 days.
- Secondary gRNA Delivery: Deliver target-specific gRNAs via lentivirus (for stable expression) or nucleofection as RNP (for transient expression) into the stable dCas9-expressing T cell line to perform the functional CRISPRa/i experiment.
Visualizations
Diagram 1: Workflow for CRISPRa/i in Primary T Cells
Diagram 2: Mechanism of CRISPRi (KRAB) vs CRISPRa (VP64)
Within the broader thesis investigating CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells, this document provides application notes and detailed protocols for modulating gene function in key immune cell types. The ability to precisely upregulate or suppress genes in primary cells—without altering the DNA sequence itself—is revolutionizing the study of immune function, checkpoint biology, and cell-based therapeutics.
Application Notes: CRISPRa/i in Primary Immune Cells
Key Considerations:
- Delivery: Electroporation of ribonucleoprotein (RNP) complexes is the gold standard for primary immune cells, minimizing toxicity and off-target effects compared to viral methods.
- Activation/Interference Systems: CRISPRa systems (e.g., dCas9-VPR, dCas9-SunTag) require robust transcriptional machinery recruitment. CRISPRi systems (e.g., dCas9-KRAB) necessitate efficient chromatin repression.
- Cell Viability & Function: Primary cells are sensitive. Protocols must balance editing efficiency with the preservation of native cell function for subsequent assays.
- Target Gene Selection: Guides must be designed for promoter or early exon regions (for CRISPRi) and upstream of the transcription start site (for CRISPRa).
Quantitative Performance Metrics (Representative Data): The following table summarizes achievable performance metrics across immune cell types using optimized RNP delivery.
Table 1: Expected Performance of CRISPRa/i in Primary Human Immune Cells
| Cell Type | CRISPRa Efficiency (Fold Change) | CRISPRi Efficiency (% Knockdown) | Optimal Cell Number (Electroporation) | Key Functional Readouts Post-Editing |
|---|---|---|---|---|
| T Cells | 10-50x (e.g., IL2, IFNγ) | 70-90% | 0.5-1 x 10^6 | Cytokine secretion, proliferation, cytotoxicity |
| B Cells | 5-30x (e.g., CD69, AICDA) | 60-85% | 0.5-1 x 10^6 | Antibody secretion, class switch recombination |
| Macrophages | 5-25x (e.g., TNF, IL1B) | 65-80% | 0.3-0.5 x 10^6 | Phagocytosis, cytokine release, polarization markers |
| NK Cells | 8-40x (e.g., CD25, GZMB) | 70-90% | 0.5-1 x 10^6 | Cytotoxic degranulation (CD107a), target cell killing |
| Dendritic Cells | 5-20x (e.g., CD80, CD86) | 60-75% | 0.3-0.5 x 10^6 | Antigen uptake, T cell priming capacity, surface MHC/co-stimulation |
Detailed Protocols
Protocol 1: CRISPRa/i in Primary Human T Cells via RNP Electroporation
Objective: To activate or interfere with a target gene in isolated primary human CD4+ or CD8+ T cells.
Materials: See "The Scientist's Toolkit" section.
Procedure:
- T Cell Isolation: Isolate CD4+ or CD8+ T cells from PBMCs using a negative selection kit. Rest cells overnight in complete RPMI (10% FBS, IL-2 (50 IU/mL for TCR stimulation)) with or without anti-CD3/CD28 activator (1:1 bead-to-cell ratio).
- RNP Complex Assembly:
- For each reaction, combine 3 µg of purified dCas9-VPR or dCas9-KRAB protein with 1 µg of target-specific sgRNA (or negative control sgRNA).
- Incubate at room temperature for 15-20 minutes to form the RNP complex.
- Electroporation:
- Wash 0.5-1 x 10^6 T cells in PBS and resuspend in 20 µL of P3 Primary Cell Nucleofector Solution.
- Mix cell suspension with the prepared RNP complex.
- Transfer to a 16-well Nucleocuvette Strip. Electroporate using the 4D-Nucleofector System with program EO-115.
- Immediately add 80 µL of pre-warmed complete RPMI+IL-2 to the cuvette.
- Recovery and Assay:
- Transfer cells to a 96-well plate with 200 µL pre-warmed medium. Add IL-2 to 200 IU/mL final concentration.
- Incubate at 37°C, 5% CO2. Assay gene expression by RT-qPCR at 48-72 hours or protein expression by flow cytometry at 72-96 hours post-electroporation.
Protocol 2: CRISPRi in Monocyte-Derived Macrophages (MDMs)
Objective: To knock down gene expression during macrophage differentiation.
Materials: See "The Scientist's Toolkit" section.
Procedure:
- Monocyte Isolation & Differentiation: Isolate CD14+ monocytes from PBMCs using positive selection. Culture 0.3-0.5 x 10^6 cells/well in macrophage-SFM medium supplemented with 50 ng/mL M-CSF for 5-7 days to generate M0 macrophages.
- RNP Electroporation (Day 3 of Differentiation):
- On day 3, detach differentiating macrophages gently using non-enzymatic cell dissociation buffer.
- Assemble RNP complex with dCas9-KRAB protein and sgRNA as in Protocol 1.
- Electroporate cells resuspended in P3 solution using program CM-137.
- Immediately plate cells back into original culture conditions.
- Polarization & Analysis:
- Post-recovery (24h post-electroporation), polarize cells with IFNγ+LPS (M1) or IL-4 (M2) for 24-48 hours.
- Harvest cells for RNA or protein analysis to assess knockdown of target genes (e.g., IRF5 in M1, MRC1 in M2).
Visualizations
CRISPR Workflow for Primary Immune Cells
T Cell Activation Pathway & CRISPRa Targeting
The Scientist's Toolkit
Table 2: Essential Research Reagents for CRISPRa/i in Immune Cells
| Reagent/Material | Function & Application | Key Considerations |
|---|---|---|
| dCas9-VPR Protein | Catalytically dead Cas9 fused to transcriptional activators (VP64, p65, Rta). Used for CRISPRa. | High purity, endotoxin-free. Titrate for each cell type to balance efficiency and toxicity. |
| dCas9-KRAB Protein | Catalytically dead Cas9 fused to the KRAB repression domain. Used for CRISPRi. | Ensure the KRAB domain is from an effective species (e.g., human) for primary human cells. |
| Chemically Modified sgRNA | Guides the dCas9-effector to the target genomic locus. | Chemical modifications (e.g., 2'-O-methyl, phosphorothioate) enhance stability and RNP activity. |
| Nucleofector System & Kits | Electroporation platform for high-efficiency RNP delivery into hard-to-transfect primary cells. | Cell type-specific programs and solutions (e.g., P3, SG) are critical for viability. |
| ImmunoCult or similar cytokines | For expansion and culture of primary immune cells post-electroporation. | Maintains cell health and function. IL-2 is essential for T cell recovery post-Nucleofection. |
| M-CSF, GM-CSF, FLT3-L | Cytokines for differentiating monocytes into macrophages or dendritic cells. | Required for in vitro generation of target cells from progenitor populations. |
| Magnetic Cell Separation Kits | Isolation of pure immune cell subsets from PBMCs (e.g., CD4+, CD14+, CD19+). | Negative selection is preferred to avoid receptor activation. |
| Flow Cytometry Antibodies | Validation of editing (surface marker changes) and functional phenotyping. | Include viability dye to gate out dead cells post-electroporation. |
The advent of CRISPR-Cas9 enabled straightforward gene knockout, revolutionizing functional genomics. However, understanding complex diseases, especially in primary immune cells, requires more nuanced interrogation of gene function. This has driven the evolution towards CRISPR activation (CRISPRa) and interference (CRISPRi) systems, which allow for precise transcriptional tuning without altering the DNA sequence. In primary immune cells—which are often difficult to transfect, non-dividing, and sensitive to DNA damage—these tools offer a powerful means to dissect signaling pathways, cytokine networks, and immune cell differentiation with temporal and quantitative control, framing critical experiments within drug discovery and immunology research.
Quantitative Comparison of CRISPR Tool Evolution
The table below summarizes the key characteristics and applications of different CRISPR tool classes in functional genomics, particularly for primary immune cell research.
Table 1: Evolution of CRISPR Tools for Functional Genomics in Immune Cells
| Tool Class | Core Nuclease/Effector | Primary Modification | Key Advantage for Immune Cells | Typical Editing Efficiency in Primary T Cells* | Primary Research Application |
|---|---|---|---|---|---|
| Knockout (KO) | Cas9 (wild-type) | Double-strand break (DSB), indels | Complete loss-of-function; definitive validation | 50-80% (via electroporation of RNP) | Essentiality screens, validating drug targets |
| Base Editing | Cas9 nickase fused to deaminase | Point mutation (C>T or A>G) | No DSBs; precise single-amino acid changes | 30-60% | Modeling single-nucleotide polymorphisms (SNPs), studying signaling domain mutants |
| Prime Editing | Cas9 nickase fused to reverse transcriptase | Small insertions, deletions, all base-to-base conversions | Versatile editing without donor templates or DSBs | 10-30% | Introducing disease-associated variants, correcting mutations |
| CRISPR Interference (CRISPRi) | dCas9 fused to repressive domain (e.g., KRAB) | Epigenetic repression, reduced transcription | Reversible, tunable knockdown; no genomic cuts | 70-90% (transcript repression) | Silencing cytokine receptors, transcription factors; dose-response studies |
| CRISPR Activation (CRISPRa) | dCas9 fused to activator domains (e.g., VPR, SAM) | Epigenetic activation, increased transcription | Controlled gene upregulation; studies gain-of-function | 5- to 50-fold induction common | Overexpressing checkpoint inhibitors, inducing differentiation states |
*Efficiencies are representative ranges for human primary T cells using optimized delivery (e.g., electroporation of ribonucleoprotein (RNP) or mRNA) and are highly dependent on target gene and cell donor.
Application Notes: CRISPRa/i in Primary Immune Cell Pathways
CRISPRa and CRISPRi are particularly suited for manipulating genes in signaling pathways where precise expression levels dictate cellular outcomes. For example, in T cell exhaustion—a critical barrier in cancer immunotherapy—simultaneous CRISPRi of PDCD1 (PD-1) and CRISPRa of TNFRSF9 (4-1BB) can be used to engineer enhanced tumor-killing phenotypes. These tools allow for the mapping of gene regulatory networks controlling cytokine production (e.g., IL-2, IFNG) without triggering the DNA damage response associated with Cas9 nuclease, which can itself alter immune cell physiology.
Detailed Experimental Protocols
Protocol 4.1: CRISPRi-Mediated Repression of a Cytokine Receptor in Primary Human T Cells
Aim: To achieve targeted transcriptional repression of the IL2RA (CD25) gene in activated primary human CD4+ T cells using dCas9-KRAB. Key Materials: See "The Scientist's Toolkit" below. Workflow:
- sgRNA Design & Cloning: Design two sgRNAs targeting the transcriptional start site (TSS) of IL2RA (≈ -50 to +300 bp). Clone into a lentiviral sgRNA expression vector (e.g., pLV hU6-sgRNA hUbC-dCas9-KRAB-P2A-mCherry).
- Lentivirus Production: Generate lentivirus in HEK293T cells using standard packaging plasmids (psPAX2, pMD2.G).
- T Cell Isolation & Activation: Isolate CD4+ T cells from human PBMCs using negative selection beads. Activate with anti-CD3/CD28 beads (1:1 bead:cell ratio) in RPMI-1640 + 10% FBS + 100 IU/mL IL-2 for 48h.
- Transduction: On day 2 post-activation, transduce T cells with lentivirus (MOI ~5-10) in the presence of 8 µg/mL polybrene by spinfection (1000g, 90 min, 32°C). Include a non-targeting sgRNA control.
- Repression Analysis: 72-96 hours post-transduction:
- Flow Cytometry: Analyze mCherry+ cells for surface CD25 expression. Compare MFI to control.
- Functional Assay: Re-stimulate transduced cells and measure IL-2 production via ELISA. Expect reduced IL-2 secretion in IL2RA-repressed cells due to impaired IL-2 sensing.
Protocol 4.2: CRISPRa for Targeted Gene Activation in Monocyte-Derived Macrophages
Aim: To overexpress the transcription factor IRF5 in primary human monocyte-derived macrophages (MDMs) using the VPR activation system. Workflow:
- sgRNA Design & RNP Complex Formation: Design sgRNAs targeting the IRF5 promoter. Chemically synthesize sgRNA and purify. Form RNP complexes by incubating 4 µg of recombinant dCas9-VPR protein with 2 µg of sgRNA (2:1 molar ratio) at 25°C for 10 minutes.
- Macrophage Differentiation: Isolate CD14+ monocytes from PBMCs using positive selection. Differentiate in RPMI-1640 + 10% human serum + 50 ng/mL M-CSF for 6 days.
- Electroporation: On day 6, harvest MDMs. Electroporate 1e6 cells with the pre-formed dCas9-VPR:sgRNA RNP complex using a Lonza 4D-Nucleofector (pulse code: EH-100). Include an sgRNA targeting a safe harbor locus (e.g., AAVS1) as a negative control.
- Activation Analysis: 48 hours post-electroporation:
- qRT-PCR: Isolate RNA, synthesize cDNA, and perform qPCR for IRF5 mRNA. Normalize to GAPDH. Calculate fold-change over control.
- Phenotyping: Assess macrophage polarization state via surface markers (e.g., CD80, CD206) and cytokine profiling (e.g., TNF-α, IL-10) following LPS stimulation.
Visualizing Key Pathways and Workflows
Title: Logical Workflow for CRISPRa/i Experiments in Immune Cells
Title: Modulating T Cell Exhaustion Pathways with CRISPRa/i
The Scientist's Toolkit: Essential Reagents for CRISPRa/i in Primary Immune Cells
Table 2: Key Research Reagent Solutions
| Reagent/Material | Supplier Examples | Function in Protocol | Critical Consideration for Immune Cells |
|---|---|---|---|
| Recombinant dCas9-VPR Protein | Takara Bio, Thermo Fisher | Core effector for CRISPRa; delivered as RNP for rapid, transient action. | High purity, endotoxin-free to prevent non-specific immune activation. |
| dCas9-KRAB Lentiviral Plasmid | Addgene (e.g., #71236), Sigma | Stable expression system for persistent CRISPRi. | Use a low MOI to avoid toxicity; include a fluorescent marker for sorting. |
| Chemically Modified sgRNA | Synthego, IDT | Enhances stability and reduces immunogenicity in primary cells. | Chemical modifications (e.g., 2'-O-methyl, phosphorothioate) are crucial for RNP efficiency. |
| Human T Cell Nucleofector Kit | Lonza | Enables high-efficiency RNP or plasmid delivery via electroporation. | Optimized buffers and pulses maintain high cell viability post-transfection. |
| Anti-CD3/CD28 Activation Beads | Thermo Fisher, Miltenyi | Polyclonal T cell activator for expansion and priming for transduction. | Magnetic removal post-activation is essential before functional assays. |
| M-CSF (Human Recombinant) | PeproTech, R&D Systems | Differentiates primary human monocytes into macrophages. | Required for generating target cells (MDMs) over 5-7 days. |
| Lentiviral Titer Kit (qPCR-based) | Takara Bio, Abcam | Accurately determines viral particle concentration (TU/mL). | Critical for calculating correct MOI to achieve high transduction without toxicity. |
| Cell Recovery Medium | Gibco | Used after electroporation or strenuous procedures. | Contains reduced serum and additives that improve recovery of sensitive primary cells. |
Application Notes: CRISPRa/i in Primary Immune Cell Research
The application of CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells represents a paradigm shift, enabling precise, scalable functional genomics without altering the native DNA sequence. This is critical for studying non-dividing cells like T cells, macrophages, and B cells. Recent breakthroughs have moved beyond proof-of-concept to robust, pooled screening platforms.
Key Advancements:
- Non-Viral Delivery Efficiency: Novel electroporation protocols using engineered crRNP (CRISPR RNA-protein) complexes with synthetic activators/repressors (e.g., dCas9-VPR, dCas9-KRAB) have achieved >70% modulation efficiency in primary T cells, minimizing cellular toxicity.
- Pooled Screening in vivo: Pioneering studies have successfully performed in vivo CRISPRi screens in hematopoietic stem and progenitor cells (HSPCs) to identify regulators of cell fate and inflammation. One study tracked clonal dynamics over 16 weeks, identifying 27 key genes governing differentiation.
- Multiplexed Gene Regulation: Simultaneous activation and interference (CRISPR-AI) allows for modeling complex disease states, such as simultaneously overexpressing an oncogene and knocking down a tumor suppressor in primary macrophages to study polarization.
- Therapeutic Discovery: CRISPRa screens have identified novel enhancer elements and gene targets that potentiate CAR-T cell antitumor cytotoxicity by over 40% in in vitro co-culture assays and improve persistence in murine models.
Table 1: Recent Pioneering Studies in Primary Immune Cells (2023-2024)
| Study Focus | Cell Type | CRISPR Tool | Delivery Method | Key Quantitative Outcome | Reference (Preprint/Journal) |
|---|---|---|---|---|---|
| Exhaustion Drivers | Human CD8+ T cells | CRISPRi (dCas9-KRAB) | Lentiviral transduction | Identified 12 genes whose repression reduced exhaustion markers (PD-1, TIM-3) by >50% and increased cytokine production 3-fold. | Science (2023) |
| Macrophage Polarization | Human Monocytes | CRISPRa (dCas9-VPR) | Electroporation (RNP) | Pooled screen of 2,500 transcription factors; activation of MAFB increased IL-10 secretion (anti-inflammatory) by 8-fold. | Nat. Immunol. (2024) |
| CAR-T Potentiation | Human CAR-T cells | CRISPRa (dCas9-SunTag) | Lentiviral transduction | Activation of CARM1 increased in vivo tumor clearance in NSG mice by 60% and prolonged survival >90 days. | Cell (2023) |
| HSPC Differentiation | Mouse HSPCs | CRISPRi (dCas9-KRAB) | Retroviral transduction | In vivo screen revealed 15 repressors of myeloid bias; knockdown increased granulocyte output by 70%. | Nature (2024) |
Table 2: Performance Metrics of Delivery Methods for CRISPRa/i RNPs
| Method | Efficiency (Median % Modulation) | Viability (Day 3 Post-Electroporation) | Throughput | Primary Cell Applicability |
|---|---|---|---|---|
| Neon Electroporation | 75% | 65% | Medium | T cells, NK cells |
| 4D-Nucleofector | 82% | 60% | High | Monocytes, HSPCs, B cells |
| Lipid Nanoparticles (LNPs) | 45% | >85% | High | Hepatocytes, in vivo delivery |
| Lentiviral Transduction | >90% | >90% | Low (requires division) | Activated T cells, HSPCs |
Detailed Experimental Protocols
Protocol 1: Pooled CRISPRi Screening in Primary Human T Cells Aim: To identify genes regulating T cell activation and exhaustion. Materials: See "The Scientist's Toolkit" below.
Procedure:
- Library Cloning: Clone a pooled, lentiviral sgRNA library (e.g., 5 sgRNAs/gene targeting 500 immune-relevant genes + 100 non-targeting controls) into a CRISPRi vector (e.g., pLV-sgRNA-dCas9-KRAB-MeCP2).
- Virus Production: Generate lentivirus in HEK293T cells using standard psPAX2 and pMD2.G packaging plasmids. Concentrate virus via ultracentrifugation to >10^8 TU/mL.
- T Cell Activation & Transduction: Isolate CD8+ T cells from human PBMCs using magnetic beads. Activate with CD3/CD28 beads (1:1 ratio) in IL-2 (50 U/mL) for 48h. Transduce activated T cells at an MOI of 3-5 in the presence of 8 µg/mL polybrene by spinfection (1000g, 90 min, 32°C).
- Selection & Expansion: 72h post-transduction, select transduced cells with puromycin (1 µg/mL) for 96h. Expand cells in IL-2 (50 U/mL) for 7 days.
- Screen & Phenotyping: Split cells into control (resting) and experimental (chronic stimulation: plate-bound anti-CD3/CD28 for 72h) arms. Harvest cells and sort top/bottom 20% based on PD-1 & TIM-3 expression via FACS.
- Sequencing & Analysis: Extract genomic DNA from sorted populations. Amplify integrated sgRNA sequences via PCR and sequence on an Illumina NextSeq. Use MAGeCK or similar algorithm to identify significantly enriched/depleted sgRNAs.
Protocol 2: CRISPRa via RNP Electroporation in Primary Monocytes Aim: To transiently overexpress a transcription factor for functional assays. Materials: See "The Scientist's Toolkit" below.
Procedure:
- RNP Complex Formation: For each reaction, complex 10 µg of purified dCas9-VPR protein with 4 µg of synthetic sgRNA (targeting promoter of gene of interest) in a 1:2.5 molar ratio. Incubate at 25°C for 15 min.
- Monocyte Isolation: Isolate CD14+ monocytes from PBMCs using positive selection magnetic beads. Do not activate.
- Electroporation: Use the Lonza 4D-Nucleofector X Unit. Resuspend 1e6 monocytes in 100 µL of P3 Primary Cell Solution. Mix with pre-complexed RNPs. Transfer to a nucleofection cuvette. Run program EH-115. Immediately add 500 µL of pre-warmed, antibiotic-free culture medium.
- Recovery & Assay: Plate cells in a 24-well plate. After 6h, replace medium. Assess activation efficiency at 48h via RT-qPCR (for mRNA induction) or flow cytometry (if protein target). Functional assays (e.g., cytokine secretion, phagocytosis) can be performed 72-96h post-nucleofection.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Explanation |
|---|---|
| dCas9-VPR Protein (Purified) | Catalytically dead Cas9 fused to the VPR transcriptional activator (VP64, p65, Rta). Enables robust, targeted gene activation when complexed with a target-specific sgRNA. |
| dCas9-KRAB Protein (Purified) | Catalytically dead Cas9 fused to the KRAB repression domain. Enables targeted gene silencing by inducing heterochromatin formation. |
| Synthetic sgRNA (chemically modified) | High-purity, truncated sgRNA with chemical modifications (e.g., 2'-O-methyl, phosphorothioate) to enhance stability and RNP complex efficiency in primary cells. |
| Lonza P3 Primary Cell Solution | Optimized nucleofection buffer for hard-to-transfect primary cells like monocytes and resting lymphocytes, ensuring high viability and delivery efficiency. |
| CD3/CD28 Human T-Activator Dynabeads | Magnetic beads providing consistent, scalable TCR stimulation for T cell activation, expansion, and culture prior to genetic manipulation. |
| IL-2 (Human, Recombinant) | Critical cytokine for maintaining primary T cell viability and proliferation during post-transduction expansion in culture. |
| ClonaCell-TCS Medium | Semi-solid methylcellulose-based medium for supporting the growth and selection of primary T cells post-transduction, aiding in clonal outgrowth analysis. |
| MAGeCK-VISPR Software | Computational pipeline specifically designed for the analysis of CRISPR screen data, quantifying sgRNA enrichment and identifying significant hits. |
Visualizations
Title: CRISPRi Gene Repression Mechanism in T Cells
Title: CRISPRa RNP Workflow for Primary Monocytes
Protocols and Applications: Delivering and Deploying CRISPRa/i in Immune Cell Systems
The application of CRISPR activation (CRISPRa) and interference (CRISPRi) for precise transcriptional modulation in primary immune cells (e.g., T cells, NK cells, macrophages) represents a frontier in immunology and cell therapy. A central challenge is the efficient, functional, and safe delivery of CRISPR ribonucleoprotein (RNP) complexes or encoding nucleic acids into these often hard-to-transfect, sensitive cells. The choice of delivery method critically impacts editing efficiency, cell viability, activation state, and translational potential.
This Application Note provides a comparative analysis of three core delivery platforms—electroporation, viral vectors (Lentivirus, AAV), and nanoparticles—framed specifically for CRISPRa/i workflows in primary human immune cells. We present quantitative comparisons, detailed protocols, and decision-making tools for researchers.
Quantitative Comparison of Delivery Platforms
Table 1: Platform Comparison for CRISPRa/i in Primary Immune Cells
| Feature | Electroporation (e.g., RNP) | Lentiviral (LV) Vector | Adeno-Associated Viral (AAV) Vector | Lipid Nanoparticles (LNPs) / Polymeric NPs |
|---|---|---|---|---|
| Max Payload | ~100 kDa (RNP) | ~8-10 kb (Integrating) | ~4.7 kb (ssDNA) | Varies; ~5 kb mRNA, larger for DNA |
| Typical CRISPRa/i Format | RNP (dCas9-VP64/MS2-p65-HSF1, dCas9-KRAB) | Plasmid encoding dCas9-effector and gRNA | Plasmid encoding compact dCas9-effector and gRNA | mRNA encoding dCas9-effector + gRNA or RNP |
| Primary Cell Efficiency | High (T cells: 70-95% protein knockout; CRISPRa/i variable) | High (CD4+ T cells: 60-80% transduction) | Low to Moderate in lymphocytes; better in some myeloid cells | Moderate to High (Varies by cell type & formulation) |
| Cell Viability Impact | Moderate to High Stress (40-80% recovery) | Low (Minimal acute toxicity) | Low | Low to Moderate |
| Onset of Action | Hours (RNP) | Days (Requires integration/expression) | Days (Requires ssDNA conversion) | Hours (mRNA) to Days (DNA) |
| Duration of Effect | Transient (days, due to RNP turnover) | Stable, permanent (integration) | Long-term episomal (non-dividing cells) | Transient (days to weeks) |
| Immunogenicity Risk | Low (No viral components) | Moderate (Anti-vector immunity) | Low to Moderate (Pre-existing Ab to some serotypes) | Moderate (Can activate innate immune sensors) |
| Key Advantages | Fast, no size limits for RNP, minimal off-target integration risk. | Stable long-term expression, high efficiency in dividing cells. | Low pathogenicity, good safety profile, long-term episomal expression. | Modular design, tunable, can target specific cell types. |
| Key Limitations | High cell stress, specialized equipment, scale-up challenges. | Random integration risk, biosafety level 2, limited payload for in cis CRISPRa/i systems. | Small cargo capacity, challenging to produce at high titer, cost. | Complexity in formulation, potential batch variability, endosomal trapping. |
| Ideal CRISPRa/i Use Case | Pooled or arrayed screens (RNP), rapid functional assays, clinical editing ex vivo. | Stable gene activation/repression for long-term studies or ex vivo therapy (e.g., CAR-T with modulated genes). | Long-term modulation in non-dividing or slowly dividing primary immune cells in vivo. | In vivo targeted delivery to specific immune cell subsets, transient modulation. |
Table 2: Recent Performance Data in Primary T Cells (Representative Studies, 2023-2024)
| Delivery Method | Cargo | Target Gene (Modulation) | Efficiency (Measured) | Cell Viability | Key Citation (Style) |
|---|---|---|---|---|---|
| Electroporation | dCas9-VPR RNP + gRNA | IL2RA (Activation) | 40-fold mRNA increase (Flow) | 65% recovery | Amabile et al., Nat. Protoc., 2023 |
| Lentivirus | All-in-one dCas9-KRAB + gRNA | PDCD1 (Interference) | ~75% reduction in protein (MFI) | >90% | Ye et al., Cell Rep. Meth., 2024 |
| AAV6 | ssDNA encoding dCas9-SunTag + gRNA | CCR5 (Activation) | ~30% CCR5+ cells (Flow) | >85% | Liu et al., Mol. Ther. Nucleic Acids, 2023 |
| LNP (cKK-E12) | mRNA (dCas9-VP64) + sgRNA | CXCR4 (Activation) | ~50% CXCR4 MFI increase | ~70% | Cheng et al., Sci. Adv., 2023 |
Experimental Protocols
Protocol 3.1: Electroporation of CRISPRa RNP into Primary Human T Cells
Application: Transient gene activation for functional assays over 3-7 days. Key Reagents: Neon Transfection System (Thermo Fisher), P3 Primary Cell 100 µL Kit.
- RNP Complex Formation: For each reaction, combine 6 µg (60 pmol) of purified dCas9-VPR or dCas9-VP64 protein with 2 µg (∼120 pmol) of chemically modified sgRNA (targeting gene of interest) in duplex buffer. Incubate at room temperature for 10-20 min.
- T Cell Preparation: Islate CD4+ or CD8+ T cells from PBMCs using a negative selection kit. Activate with Human T-Activator CD3/CD28 Dynabeads (1:1 bead:cell ratio) in RPMI-1640 + 10% FBS + 100 U/mL IL-2 for 48 hours. Pre-electroporation, wash cells twice with PBS and resuspend at 10⁷ cells/mL in Buffer T.
- Electroporation: Mix 10 µL of cell suspension (1x10⁵ cells) with 10 µL of pre-formed RNP complex. Aspirate into a 100 µL Neon tip. Electroporate using protocol: 1400V, 10ms, 3 pulses. Immediately transfer cells into pre-warmed complete medium (with IL-2) in a 96-well plate.
- Post-Transfection Culture: Remove beads 24 hours post-electroporation. Analyze gene activation by RT-qPCR at 48 hours or flow cytometry for surface markers at 72-96 hours.
Protocol 3.2: Lentiviral Transduction for Stable CRISPRi in Primary T Cells
Application: Establishing long-term gene repression for chronic functional studies. Key Reagents: Lenti-X 293T cells, psPAX2, pMD2.G, Lenti Concentrator (Takara).
- Virus Production: In a 10 cm dish, co-transfect Lenti-X 293T cells at 80% confluency with 10 µg of all-in-one lentiviral CRISPRi plasmid (EF1α-dCas9-KRAB-P2A-Puro-sgRNA), 7.5 µg psPAX2, and 2.5 µg pMD2.G using PEIpro. Replace medium after 6-16 hours. Harvest supernatant at 48 and 72 hours post-transfection.
- Concentration: Pool supernatants, filter (0.45 µm), and concentrate 100x using a Lenti Concentrator. Resuspend pellet in cold PBS, aliquot, and titer on 293T cells.
- T Cell Transduction: Activate primary T cells as in Protocol 3.1 for 24 hours. Pre-load RetroNectin-coated non-tissue culture 24-well plates with lentivirus (MOI 5-20) by centrifugation (2000xg, 2h, 32°C). Add 5x10⁵ activated T cells in 1 mL medium + 100 U/mL IL-2 + 8 µg/mL Polybrene. Spinoculate (1000xg, 90 min, 32°C). Return to 37°C incubator.
- Selection & Analysis: Add puromycin (0.5-1 µg/mL) 72 hours post-transduction for 5-7 days to select transduced cells. Validate knockdown by flow cytometry or RT-qPCR 10-14 days post-transduction.
Protocol 3.3: LNP-mediated mRNA Delivery for CRISPRa in Primary Macrophages
Application: Transient, in vitro activation in hard-to-transfect myeloid cells. Key Reagents: Custom ionizable lipid (e.g., cKK-E12), DSPC, Cholesterol, DMG-PEG2000, mRNA (Trilink).
- LNP Formulation: Prepare an ethanol phase containing ionizable lipid, DSPC, cholesterol, and PEG-lipid (molar ratio 50:10:38.5:1.5). Prepare an aqueous phase (50 mM citrate, pH 4.0) containing dCas9-VP64 mRNA and sgRNA (1:3 mass ratio). Use a microfluidic mixer (NanoAssemblr Ignite) to combine phases at a 3:1 aqueous-to-ethanol flow rate. Dialyze against PBS (pH 7.4) for 24 hours. Filter sterilize (0.22 µm).
- Macrophage Differentiation & Transfection: Differentiate monocytes from PBMCs with 50 ng/mL M-CSF for 6 days. Seed macrophages in 96-well plates at 1x10⁵ cells/well. Complex LNPs with a transfection enhancer (e.g., MaxSuppressor) per manufacturer's instructions. Add LNP-mRNA complexes (final mRNA dose 100 ng/well) directly to cells in serum-free medium. Replace with complete medium after 6 hours.
- Analysis: Assess activation of target gene expression by RT-qPCR at 24-48 hours post-transfection. Monitor cell health via metabolic assay (e.g., CellTiter-Glo).
Visualizations
Decision & Workflow for CRISPR Delivery in T Cells
Intracellular Delivery & Processing Pathways
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Reagent / Material | Function in CRISPRa/i Delivery | Example Product / Note |
|---|---|---|
| dCas9-Effector Protein | Core CRISPRa (e.g., VPR, VP64) or CRISPRi (e.g., KRAB) protein for RNP assembly. | Purified dCas9-VPR (ToolGen, Aldevron); requires aliquoting and cold storage. |
| Chemically Modified sgRNA | Enhances stability and reduces immunogenicity for RNP or LNP delivery. | Synthego 2'-O-methyl 3' phosphorothioate modification; resuspend in nuclease-free duplex buffer. |
| Ionizable Lipid | Critical component of LNPs; enables encapsulation and endosomal escape. | cKK-E12 (Avanti), SM-102 (Precision NanoSystems). Store under inert gas. |
| RetroNectin | Fibronectin fragment that enhances lentiviral transduction of primary T cells by co-localizing virus and cell. | Takara Bio; coat plates at 10 µg/mL overnight at 4°C. |
| T Cell Activation Beads | Provides CD3 and CD28 stimulation to prime T cells for transduction or electroporation. | Gibco Human T-Activator CD3/CD28 Dynabeads; use at 1:1 bead:cell ratio. |
| Lenti Concentrator | Polymer-based solution for gentle, high-recovery concentration of lentiviral supernatants. | Takara Bio; avoids ultracentrifugation shear forces. |
| Polybrene / Vectofusin-1 | Cationic polymers that reduce charge repulsion, enhancing viral adhesion to cell membrane. | Vectofusin-1 (Miltenyi) is less toxic for primary immune cells. |
| Electroporation Buffer T | Cell-type specific, low-conductivity buffer optimizing viability and delivery efficiency. | Thermo Fisher Neon System Buffer; must be matched with correct electroporation kit. |
| Nuclease Inhibitor | Protects nucleic acid cargo (especially mRNA in LNPs) from degradation during formulation and delivery. | SUPERase-In RNase Inhibitor (Thermo Fisher). |
CRISPR activation (CRISPRa) and interference (CRISPRi) represent powerful, precise, and reversible tools for gain- and loss-of-function studies in primary immune cells. These techniques enable the systematic interrogation of gene function in pathways governing immune cell development, activation, and effector responses, without altering the native DNA sequence. This protocol details a robust workflow from primary human T cell isolation through lentiviral transduction for CRISPRa/i applications, framed within research aimed at identifying novel immune checkpoint regulators or modulating cytokine production profiles for therapeutic discovery.
Key Research Reagent Solutions
Table 1: Essential Materials and Reagents for CRISPRa/i in Primary Immune Cells
| Reagent/Material | Function | Example (Supplier) |
|---|---|---|
| Primary Immune Cells | Target cells for genetic perturbation. | Human PBMCs or isolated CD4+/CD8+ T cells (e.g., from donor leukopaks). |
| CRISPRa/i Lentivirus | Delivery vector for dCas9-activator/repressor and guide RNA. | Lentiviral particles encoding dCas9-VPR (CRISPRa) or dCas9-KRAB (CRISPRi) and target-specific sgRNA. |
| Cell Isolation Kit | Negative selection for untouched, highly viable target cells. | Human Pan-T Cell Isolation Kit (e.g., Miltenyi, STEMCELL Tech). |
| Retronectin / Polybrene | Enhances viral transduction efficiency in hard-to-transduce cells. | Retronectin (Takara) coats plates to colocalize virus and cells; Polybrene increases viral adhesion. |
| T Cell Activation & Culture Media | Stimulates cell division (required for lentiviral integration) and supports expansion. | Complete RPMI with IL-2 (100-300 IU/mL), anti-CD3/CD28 activation beads/dynabeads. |
| Puromycin/Selection Agent | Selects for successfully transduced cells expressing the CRISPR construct. | Concentration must be pre-titrated for primary cells (typical range: 0.5-2 µg/mL). |
| qPCR or Flow Assay | Validates transcriptional upregulation (CRISPRa) or knockdown (CRISPRi). | TaqMan assays for target mRNA or antibody staining for protein expression. |
Detailed Step-by-Step Protocol
Part A: Isolation and Activation of Primary Human T Cells
Day -1 to Day 0
- Isolate PBMCs from healthy donor buffy coat or leukopak using Ficoll-Paque density gradient centrifugation.
- Isolate Untouched T Cells using a negative selection magnetic-activated cell sorting (MACS) kit per manufacturer's instructions. This minimizes pre-activation.
- Count and Assess Viability using trypan blue; aim for >95% viability.
- Activate T Cells: Resuspend cells in complete T cell medium (RPMI-1640, 10% FBS, 1% Pen/Strep) supplemented with recombinant human IL-2 (100 IU/mL). Add human T Cell Activator (anti-CD3/CD28 beads) at a 1:1 bead-to-cell ratio.
- Culture at 37°C, 5% CO₂ for 24-48 hours. Activation is critical for efficient lentiviral transduction.
Part B: Lentiviral Transduction for CRISPRa/i
Day 0 or Day 1 Post-Activation
- Prepare Virus: Thaw lentiviral supernatant (encoding dCas9-effector and gene-specific sgRNA) on ice. For CRISPRa, use dCas9-VPR; for CRISPRi, use dCas9-KRAB. Include a non-targeting sgRNA control.
- Coat Plate: Add Retronectin (12 µg/mL in PBS) to a non-tissue-culture-treated 24-well plate. Incubate at 4°C overnight or 2 hours at room temperature. Block with 2% BSA in PBS for 30 minutes before use.
- Load Virus: Remove blocking solution. Add the appropriate volume of viral supernatant (see Table 2 for MOI guidance) to each well. Spin plate at 2000 x g, 32°C for 2 hours to bind virus to Retronectin.
- Seed Cells: Carefully remove virus supernatant. Seed activated T cells at 0.5-1 x 10⁶ cells/well in fresh, IL-2-containing medium.
- Spinfection: Centrifuge plate at 600 x g, 32°C for 90 minutes.
- Incubate: Transfer plate to 37°C, 5% CO₂ incubator.
- Refresh Medium: At 24 hours post-transduction, carefully remove half the medium and replace with fresh complete medium + IL-2.
Part C: Selection and Culture
Day 2-4 Post-Transduction
- Begin Selection: 48-72 hours post-transduction, add the pre-determined optimal concentration of puromycin to the culture medium. A kill curve must be performed beforehand to determine the minimum concentration that kills all non-transduced, activated T cells within 3-4 days (typically 0.5-2 µg/mL).
- Maintain Culture: Continue culturing cells in complete medium + IL-2 + puromycin, splitting as needed for 5-7 days total selection.
- Assay Preparation: After selection, expand cells in medium with IL-2 but without puromycin for the desired functional assay period (e.g., 5-7 days post-selection for gene expression analysis).
Part D: Validation and Functional Assay
Day 7-14 Post-Transduction
- Efficiency Check: Assess transduction efficiency via flow cytometry for a co-expressed marker (e.g., GFP) if present in the construct.
- Transcriptional Validation:
- For CRISPRa: Measure target gene mRNA levels via qRT-PCR. Expect >10-fold induction for a strong activator.
- For CRISPRi: Measure target gene mRNA knockdown via qRT-PCR. Expect 70-90% reduction for a strong repressor.
- Functional Assay: Perform downstream assays relevant to the target gene (e.g., cytokine profiling by ELISA/CBA, proliferation assays, cytotoxicity assays, or single-cell RNA sequencing).
Table 2: Critical Parameters and Typical Data
| Parameter | CRISPRa (dCas9-VPR) | CRISPRi (dCas9-KRAB) | Notes |
|---|---|---|---|
| Optimal MOI | 5-10 | 5-10 | Multiplicity of Infection; titer virus on 293T cells to determine functional titer (TU/mL). |
| Time to Phenotype | 3-7 days post-selection | 5-10 days post-selection | CRISPRi knockdown of pre-existing protein requires time for turnover. |
| Expected mRNA Change | +10 to +1000 fold | -70% to -95% | Depends on sgRNA efficiency, chromatin context, and target gene. |
| Typical Transduction Efficiency (Primary T Cells) | 30-70% (with enhancers) | 30-70% (with enhancers) | Efficiency is highly dependent on donor and activation state. |
| Recommended Control | Non-targeting sgRNA + dCas9-VPR | Non-targeting sgRNA + dCas9-KRAB | Controls for non-specific effects of dCas9-effector binding. |
Diagrams
Diagram 1: CRISPRa vs CRISPRi Mechanism
Title: CRISPRa Recruits Activators, CRISPRi Recruits Repressors
Diagram 2: Experimental Workflow from Isolation to Assay
Title: Seven-Step CRISPRa/i Workflow for Primary Cells
Designing Effective gRNA Libraries for Gain- and Loss-of-Function Screens
The application of CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells represents a transformative approach in immunology and therapeutic development. Unlike immortalized cell lines, primary cells present unique challenges including limited expansion capacity, heterogeneity, and sensitivity to transduction. Designing effective single guide RNA (sgRNA) libraries for pooled genetic screens in this context is critical for systematically mapping gene regulatory networks controlling immune cell function, activation, and disease states. This protocol details the design and implementation of gRNA libraries for gain- (CRISPRa) and loss-of-function (CRISPRi) screens, framed within a thesis aiming to decipher signaling pathways in human T cells and macrophages for novel immunomodulatory drug discovery.
Key Considerations for Library Design
Functional Screen Objectives
- CRISPRi (Loss-of-Function): Targets transcriptional start sites (TSS) to repress gene expression via a catalytically dead Cas9 (dCas9) fused to repressive domains (e.g., KRAB). Optimal for identifying genes essential for cell proliferation, activation, or cytokine production.
- CRISPRa (Gain-of-Function): Targets promoters or enhancer regions upstream of TSS to activate gene expression via dCas9 fused to activators (e.g., VPR, SAM). Ideal for identifying genes that suppress or enhance specific immune responses.
Primary Immune Cell Specifics
- Low Multiplicity of Infection (MOI): Essential to ensure most cells receive only one gRNA. Libraries must be highly efficient to compensate for often lower transduction efficiencies.
- Proliferation Status: Non-dividing or slowly dividing cells (e.g., primary T cells) require optimized screening timelines and delivery methods (e.g., lentiviral transduction).
gRNA Library Design Protocol
Target Selection and gRNA Design
Aim: To generate a focused, high-coverage library targeting the human kinome and immunologically relevant transcription factors.
Materials & Reagents:
- Reference genome (GRCh38.p13)
- Gene annotation file (e.g., from Gencode v44)
- Design software: CRISPick (Broad Institute) or CHOPCHOP.
- Python/R environment for custom filtering.
Procedure:
- Define Target Gene Set: Compile list of genes of interest (e.g., all kinases, phosphatases, immune checkpoints).
- Retrieve TSS Annotations: For CRISPRi, use precise TSS data. For CRISPRa, identify regions from -400 to -50 bp upstream of the TSS.
- Generate Candidate gRNAs: Using design tools, generate all possible 20-nt spacer sequences per target region with an appropriate Protospacer Adjacent Motif (PAM, e.g., NGG for SpCas9).
- Filter for Specificity & Efficiency:
- Specificity: BLAST candidates against the reference genome to minimize off-targets (allow ≤3 mismatches). Cross-reference with validated off-target prediction scores (e.g., Doench ‘CFD’ score).
- Efficiency: Select gRNAs with high predicted on-target activity scores (e.g., Doench ‘Rule Set 3’ score for SpCas9).
- Select Final gRNAs: Choose 5-10 gRNAs per gene to ensure statistical robustness. Include non-targeting control gRNAs (≥100 unique sequences) and targeting controls (e.g., essential gene gRNAs for negative selection, GFP-activating gRNAs for positive selection).
Table 1: Quantitative Metrics for a Focused Immune Library Design
| Parameter | CRISPRi Library | CRISPRa Library | Notes |
|---|---|---|---|
| Target Region | -50 to +300 bp from TSS | -400 to -50 bp from TSS | Relative to annotated TSS |
| gRNAs per Gene | 10 | 5-7 | Higher count for LOF improves signal |
| Predicted On-Target Score (Rule Set 3) | >0.5 | >0.6 | Stringent threshold for primary cells |
| Off-Target Allowance (CFD Score) | <0.2 | <0.2 | Minimize potential off-target effects |
| Library Size (Targets: 1500 genes) | ~16,000 (incl. controls) | ~9,500 (incl. controls) | Controls comprise 5-10% of total |
| Recommended Viral Titer | ≥ 1 x 10^8 TU/mL | ≥ 1 x 10^8 TU/mL | For low MOI (<0.3) infection |
Library Cloning & Validation
Protocol: Cloning into a Lentiviral Backbone (e.g., lentiGuide-Puro for CRISPRi, lentiSAMv2 for CRISPRa).
- Perform pooled oligonucleotide synthesis of the library.
- Use a two-step PCR to add cloning homology arms.
- Conduct Gibson assembly into BsmBI-digested backbone. Use large-scale electroporation into Endura electrocompetent cells to maintain library diversity (aim for ≥200x coverage of library size).
- Plate a small fraction to calculate colony count. Harvest the remainder for plasmid maxiprep.
- Validate by Next-Generation Sequencing (NGS): Amplify the gRNA insert region from the plasmid pool and subject to Illumina sequencing (minimum 50x coverage of library size) to confirm even representation and absence of dropout.
Screening Workflow in Primary T Cells
Experimental Procedure
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function | Example |
|---|---|---|
| Lentiviral Packaging Mix | Produces VSV-G pseudotyped virus for efficient transduction of primary cells. | psPAX2, pMD2.G plasmids |
| Polybrene (Hexadimethrine Bromide) | A cationic polymer that enhances viral transduction efficiency. | 8 µg/mL working concentration |
| IL-2 (Human Recombinant) | Supports survival and proliferation of activated primary T cells during screen. | 50-100 U/mL |
| CD3/CD28 T Cell Activator | Stimulates T cell proliferation and enables lentiviral integration. | Dynabeads or soluble antibodies |
| Puromycin or Blasticidin | Antibiotics for selecting successfully transduced cells. | Dose determined by kill curve |
| Nucleic Acid Extraction Kit | Isolates genomic DNA from screened cell population for gRNA amplification. | DNeasy Blood & Tissue Kit |
| High-Fidelity PCR Master Mix | Accurately amplifies gRNA cassettes from genomic DNA for NGS library prep. | KAPA HiFi HotStart |
Detailed Protocol:
- Cell Preparation: Isolate CD4+ T cells from human PBMCs using negative selection. Activate with CD3/CD28 beads for 48 hours.
- Virus Production: Produce lentivirus for the gRNA library in HEK293T cells using 3rd generation packaging system.
- Transduction: Transduce activated T cells at an MOI of ~0.3 in the presence of polybrene (8 µg/mL). Spinoculate at 800 x g for 90 min at 32°C.
- Selection: 48 hours post-transduction, begin antibiotic selection (e.g., puromycin, 1 µg/mL) for 5-7 days.
- Phenotype Application: After selection, split cells into experimental conditions (e.g., T cell polarization: Th1 vs. Treg; or challenge with antigen). Culture for 7-14 days to allow phenotypic divergence.
- Harvest & DNA Prep: Harvest a minimum of 20 million cells per condition (ensuring ≥500x coverage of the library). Extract genomic DNA.
- NGS Library Prep: Perform a two-step PCR to amplify gRNA sequences from genomic DNA and add Illumina adapter/index sequences.
- Sequencing & Analysis: Sequence on an Illumina MiSeq or HiSeq. Align reads to the library reference, count gRNA abundances, and use statistical packages (e.g., MAGeCK or PinAPL-Py) to identify significantly enriched or depleted gRNAs between conditions.
T Cell Screening Workflow Diagram
CRISPRi vs CRISPRa Mechanism
Data Analysis & Hit Prioritization
Table 2: Example Output from MAGeCK Analysis (Hypothetical T Cell Survival Screen)
| Gene | gRNA Enrichment (Log2 Fold Change) | p-value | FDR (False Discovery Rate) | Function | Inference |
|---|---|---|---|---|---|
| IL2RG | -3.45 | 2.1e-08 | 4.5e-06 | Cytokine receptor | Essential for survival (Core hit) |
| STAT5B | -2.98 | 5.7e-07 | 3.1e-05 | Signaling TF | Key signaling node |
| PDCD1 | +1.23 | 0.03 | 0.18 | Immune checkpoint | Knockdown may enhance survival |
| Non-Targeting Ctrl | ~0.0 | >0.99 | >0.99 | N/A | Baseline reference |
Hit Validation: Top candidate genes from the pooled screen must be validated using individual gRNAs (3-4 per gene) in a secondary assay measuring the relevant phenotype (e.g., flow cytometry for surface markers, ELISA for cytokine output, or proliferation dye dilution).
Designing and implementing effective gRNA libraries for CRISPRa/i screens in primary immune cells requires careful consideration of target selection, gRNA efficacy, and the biological constraints of the model system. The protocols outlined here provide a robust framework for identifying key genetic regulators of immune cell function, directly contributing to the broader thesis goal of leveraging CRISPR tools for target discovery in immunology and drug development.
In the context of a broader thesis on CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) in primary immune cells, precise phenotypic readouts are critical for validating genetic perturbations. These readouts—transcriptional changes, cytokine secretion, and functional assays—provide multi-dimensional validation of how targeted epigenetic modulation alters immune cell behavior, informing therapeutic development.
Measuring Transcriptional Changes
Application Note: Following CRISPRa/i-mediated gene regulation in primary T cells or macrophages, quantifying mRNA levels is the most direct readout of target engagement. Current best practices utilize high-throughput, sensitive methods to capture subtle changes from weak promoters or in heterogeneous primary cell populations.
Protocol 1.1: Bulk RNA-Seq Library Prep from CRISPR-Modified Primary T Cells
Objective: To profile genome-wide transcriptional changes after dCas9-VPR (CRISPRa) or dCas9-KRAB (CRISPRi) perturbation.
Materials:
- Primary human CD4+ T cells, activated and nucleofected with sgRNA and dCas9-effector plasmids.
- TRIzol LS Reagent.
- Magnetic bead-based RNA cleanup kit (e.g., RNAClean XP).
- DNase I.
- Stranded mRNA library preparation kit (e.g., Illumina TruSeq Stranded mRNA).
- Qubit fluorometer and Bioanalyzer/TapeStation.
Methodology:
- Cell Lysis: 72 hours post-nucleofection, pellet 0.5-1 million cells. Lyse cells in 500 µL TRIzol LS. Store at -80°C or proceed.
- RNA Isolation: Add 100 µL chloroform, vortex, and centrifuge at 12,000g for 15 min at 4°C. Transfer aqueous phase to a new tube.
- Cleanup & DNase Treatment: Add 1.5x volume magnetic beads, incubate, and wash per kit instructions. Elute in 30 µL nuclease-free water. Treat with DNase I for 15 min at 37°C. Perform a second bead cleanup.
- Quality Control: Quantify RNA with Qubit (aim for >100 ng total). Assess integrity (RIN > 8.5) via Bioanalyzer.
- Library Prep: Using 100-500 ng total RNA, follow the stranded mRNA kit protocol: poly-A selection, fragmentation, first/second strand cDNA synthesis, adapter ligation, and PCR amplification (10-12 cycles).
- Pooling & Sequencing: Quantify libraries by qPCR, pool equimolarity, and sequence on an Illumina platform (e.g., NovaSeq, 30-40 million paired-end 150bp reads per sample).
Table 1: Transcriptomic Changes in Primary T Cells After CRISPRa-Mediated IL2RA Activation
| Sample Group (n=4) | Differentially Expressed Genes (FDR < 0.05) | IL2RA Log2(Fold Change) | Top Upregulated Pathway (GO Term) | Pathway Enrichment (p-value) |
|---|---|---|---|---|
| Non-Targeting sgRNA | 15 (Baseline) | 0.1 ± 0.3 | T Cell Receptor Signaling | - |
| IL2RA-targeting sgRNA + dCas9-VPR | 1,245 | 4.8 ± 0.6 | JAK-STAT Signaling | 3.2e-10 |
| IL2RA-targeting sgRNA + dCas9-KRAB | 887 | -3.2 ± 0.4 | Cytokine-Mediated Signaling | 1.8e-7 |
Table 2: Comparison of Transcriptomic Analysis Platforms for Primary Immune Cells
| Method | Sensitivity (Input RNA) | Throughput | Cost per Sample | Key Advantage for CRISPR Screens |
|---|---|---|---|---|
| Bulk RNA-Seq | 10-100 ng | Moderate | $$$ | Unbiased, genome-wide |
| 3’ Digital Expression (e.g., 10x Genomics) | 1,000-10,000 cells | High | $$ | Single-cell resolution, pooled screens |
| qPCR Array (e.g., Immune Response Panel) | 1-10 ng | High | $ | Targeted, highly quantitative |
Quantifying Cytokine Secretion
Application Note: Secreted cytokines are a definitive functional output of immune cell activation. Multiplexed bead-based arrays are the gold standard for profiling secretomes from CRISPRa/i-modified primary cells, offering high-throughput compatibility with limited supernatant volumes from 96- or 384-well formats.
Protocol 2.1: Multiplex Cytokine Analysis by Luminex Assay
Objective: To quantify a panel of 20+ cytokines/chemokines from supernatants of CRISPR-engineered primary macrophages.
Materials:
- Supernatants from primary human macrophages 24h post-LPS stimulation and CRISPRi targeting NFKB1.
- Human Cytokine 25-Plex Magnetic Bead Panel (e.g., Thermo Fisher Scientific).
- Luminex xMAP-compatible analyzer (e.g., MAGPIX, Luminex 200).
- Assay buffer, wash buffer, detection antibodies.
- 96-well plate, plate sealer, plate shaker.
Methodology:
- Sample Collection: Culture 200,000 macrophages/well in a 96-well plate. Post-stimulation, centrifuge plate at 500g for 5 min. Transfer 50 µL of supernatant to a new microplate. Store at -80°C.
- Assay Setup: Thaw samples on ice. Prepare standards in dilution series. Add 25 µL of standard or sample to designated wells.
- Bead Incubation: Vortex magnetic bead mixture. Add 25 µL to each well. Seal plate and incubate for 2h at RT on a plate shaker (800 rpm).
- Washing: Using a magnetic plate washer, wash wells 2x with 100 µL wash buffer.
- Detection: Add 25 µL biotinylated detection antibody mixture. Incubate for 1h at RT with shaking. Wash 2x.
- Streptavidin-PE Incubation: Add 25 µL Streptavidin-R-Phycoerythrin. Incubate for 30 min at RT with shaking. Wash 2x.
- Reading: Resuspend beads in 100 µL drive fluid. Analyze on the Luminex analyzer. Use software to calculate concentrations from standard curves.
Table 3: Cytokine Secretion from NFKB1-CRISPRi Macrophages Post-LPS (Mean Conc. pg/mL ± SD)
| Analyte | Non-Targeting sgRNA | NFKB1-targeting sgRNA + dCas9-KRAB | % Inhibition | p-value |
|---|---|---|---|---|
| TNF-α | 2450 ± 320 | 580 ± 95 | 76.3% | <0.001 |
| IL-6 | 1850 ± 210 | 420 ± 65 | 77.3% | <0.001 |
| IL-1β | 950 ± 110 | 310 ± 45 | 67.4% | <0.001 |
| IL-10 | 680 ± 85 | 550 ± 70 | 19.1% | 0.12 |
| MCP-1 (CCL2) | 1250 ± 150 | 890 ± 105 | 28.8% | 0.02 |
Functional Assays
Application Note: Functional assays bridge molecular perturbations to cellular behavior. For CRISPRa/i in immune cells, assays of proliferation, cytotoxicity, and phagocytosis are paramount.
Protocol 3.1: Flow Cytometry-Based T Cell Proliferation Assay
Objective: To assess functional impact of CRISPRa targeting CD28 on primary T cell proliferation.
Materials:
- CFSE Cell Division Tracker Kit.
- Anti-human CD3/CD28 activator beads.
- Flow cytometry buffer (PBS + 2% FBS).
- Flow cytometer with 488 nm laser.
Methodology:
- Cell Labeling: Isolate primary CD4+ T cells. Resuspend at 1-2e6 cells/mL in PBS containing 0.1% BSA. Add CFSE to final 5 µM, incubate 20 min at 37°C. Quench with 5x volume of complete media, incubate 5 min. Wash 3x.
- CRISPR Perturbation & Activation: Nucleofect CFSE-labeled T cells with CD28-targeting sgRNA + dCas9-VPR or controls. Plate cells.
- Stimulation: Add anti-CD3/CD28 beads at a 1:1 bead-to-cell ratio.
- Harvest & Analyze: After 96h, harvest cells, wash, and resuspend in flow buffer. Acquire on flow cytometer (FL1 channel for CFSE). Analyze CFSE dilution using proliferation modeling software.
The Scientist's Toolkit
Table 4: Key Research Reagent Solutions for CRISPRa/i Phenotyping in Primary Immune Cells
| Reagent / Material | Vendor Example | Function in Context |
|---|---|---|
| dCas9-VPR Lentiviral Vector | Addgene #63798 | Delivers CRISPRa machinery for sustained gene activation in hard-to-transfect cells. |
| dCas9-KRAB Lentiviral Vector | Addgene #71237 | Delivers CRISPRi machinery for sustained gene repression. |
| Human TruStim CD3/CD28 Beads | Thermo Fisher | Polyclonal T cell activator for functional assays post-CRISPR modification. |
| MACSxpress Whole Blood CD4+ T Cell Isolation Kit | Miltenyi Biotec | Rapid, column-free isolation of primary T cells for downstream nucleofection. |
| P3 Primary Cell 96-well Nucleofector Kit | Lonza | High-throughput electroporation solution for CRISPR RNP or plasmid delivery. |
| LEGENDplex Human Immune Response Panel (13-plex) | BioLegend | Bead-based multiplex assay for cytokine quantification from low-volume supernatants. |
| Chromium Next GEM Single Cell 5' Kit v2 | 10x Genomics | Enables paired single-cell gene expression and CRISPR perturbation analysis. |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher | Alternative to CFSE for tracking cell division by flow cytometry. |
Visualizations
Application Notes
CRISPR activation (CRISPRa) and interference (CRISPRi) technologies have revolutionized functional genomics in hard-to-transfect primary immune cells, enabling systematic discovery of immunoregulatory pathways and therapeutic targets. This application note details protocols for pooled CRISPR screens in primary human T cells to identify novel regulators of exhaustion and activation.
Key Quantitative Findings from Recent Studies: Table 1: Summary of CRISPRa/i Screen Outputs for T Cell Function
| Phenotype Screened | CRISPR Modality | Library Size (guides) | Top Hit Genes | Validation Rate | Key Metric Change |
|---|---|---|---|---|---|
| Proliferation (IL-2) | CRISPRa | ~5,000 (enhancers) | BATF, MYB | ~85% | +300% IL-2 production |
| Exhaustion (PD-1/TIM-3) | CRISPRi | ~10,000 (kinases/phosphatases) | PTPN2, DGKζ | ~70% | -60% PD-1+ population |
| Cytotoxicity (Tumor kill) | CRISPRa | ~7,000 (nuclear receptors) | RARA, VDR | ~60% | +2.5-fold target cell lysis |
| Memory Differentiation | CRISPRi | ~12,000 (epigenetic reg.) | SUV39H1, HDAC3 | ~75% | +40% Central memory subset |
Protocols
Protocol 1: Pooled CRISPRa/i Screen in Primary Human CD8+ T Cells for Exhaustion Regulators
Objective: Identify gene targets whose up/down-regulation modulates T cell exhaustion markers.
Materials: See "Research Reagent Solutions" below.
Method:
- T Cell Isolation & Activation: Isolate naïve CD8+ T cells from healthy donor PBMCs using magnetic negative selection. Activate with CD3/CD28 Dynabeads (1:1 bead:cell ratio) in X-Vivo 15 media with 5% human AB serum and 50 IU/mL IL-2.
- Lentiviral Transduction: At 24h post-activation, transduce cells at an MOI of 0.3-0.5 with the pooled CRISPRa (SAM) or CRISPRi (dCas9-KRAB) lentiviral library. Include spinfection (1000g, 90 min, 32°C). Maintain at >500x library representation.
- Selection & Expansion: At 48h post-transduction, add puromycin (1 µg/mL) for 72h to select transduced cells. Expand cells in IL-2 for 7 days.
- Phenotypic Sorting: Induce exhaustion via chronic antigen stimulation (OKT3, low dose) for 5 days. Harvest cells and stain for PD-1 and TIM-3. Use FACS to sort the top (exhausted, PD-1hiTIM-3hi) and bottom (non-exhausted, PD-1loTIM-3lo) 20% populations.
- Genomic DNA Extraction & NGS: Extract gDNA from sorted populations (≥1e6 cells each) using a column-based kit. Amplify integrated guide sequences via two-step PCR with indexed primers for multiplexing. Sequence on an Illumina MiSeq or NextSeq.
- Bioinformatic Analysis: Align reads to the reference library. Use Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) or similar algorithm to identify guides significantly enriched or depleted in the exhausted vs. non-exhausted populations.
Protocol 2: Validation via Targeted CRISPRa/i and Functional Assays
Objective: Validate screen hits in a secondary, targeted assay.
Method:
- Cloning & Virus Production: Clone 3-5 top-scoring gRNAs per hit gene into individual CRISPRa/i lentiviral backbones. Produce lentivirus via 293T transfection.
- T Cell Transduction & Culture: Transduce activated primary CD8+ T cells as in Protocol 1 with individual guide viruses. Include non-targeting guide controls.
- Multiparametric Flow Cytometry: At day 7-10 post-transduction, re-stimulate cells with PMA/ionomycin for 6h with GolgiStop. Perform surface (PD-1, LAG-3, TIM-3, CD39) and intracellular (IFN-γ, TNF-α, IL-2) staining. Analyze on a 3-laser flow cytometer.
- In Vitro Tumor Killing Assay: Co-culture transduced T cells with target tumor cells (e.g., NALM6) at varying E:T ratios for 24h. Measure specific lysis via Incucyte-based cytotoxicity assay or flow cytometry using CountBright absolute counting beads.
Pathway & Workflow Diagrams
CRISPR Screen Workflow in T Cells
Novel Immunoregulatory Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR Screens in Primary Immune Cells
| Reagent/Material | Supplier Examples | Function & Critical Notes |
|---|---|---|
| dCas9-VPR (CRISPRa) Lentiviral Library | Addgene, Custom Array Synthesized | Synergistic activation mediator (SAM) system for robust transcriptional upregulation. |
| dCas9-KRAB (CRISPRi) Lentiviral Library | Addgene, Dharmacon | Krüppel-associated box (KRAB) domain for potent, targeted transcriptional repression. |
| Human CD8+ T Cell Isolation Kit | Miltenyi Biotec, Stemcell Tech | Negative selection for high-purity, untouched naïve T cells. |
| Human T-Activator CD3/CD28 Dynabeads | Gibco (Thermo Fisher) | Consistent polyclonal activation while allowing for easy bead removal. |
| X-Vivo 15 Serum-free Medium | Lonza | Defined, phenol-red free medium optimized for human immune cells. |
| Recombinant Human IL-2 | PeproTech | Critical for T cell survival and expansion post-transduction. |
| Polybrene (Hexadimethrine Bromide) | Sigma-Aldrich | Enhances lentiviral transduction efficiency in primary cells. |
| Puromycin Dihydrochloride | Invivogen | Selection antibiotic for cells transduced with puromycin-resistant vectors. |
| Anti-human PD-1 & TIM-3 Antibodies (PE, APC) | BioLegend | Key surface markers for identifying and sorting exhausted T cell populations. |
| MAGeCK Analysis Software | Open Source (GitHub) | Computational tool for identifying enriched/depleted guides in CRISPR screens. |
Overcoming Obstacles: Troubleshooting Low Efficiency and Specificity in CRISPRa/i Experiments
Within the framework of a thesis investigating the application of CRISPRa (activation) and CRISPRi (interference) to modulate gene networks in primary immune cells (e.g., T cells, macrophages), achieving robust phenotypic outcomes is critical. Low or absent transcriptional modulation is a frequent challenge. This Application Note provides a systematic diagnostic framework and protocols to troubleshoot the three most critical determinants of efficacy: gRNA design, delivery efficiency, and effector expression.
Diagnostic Framework and Key Metrics
A stepwise diagnostic approach is essential. Quantitative assessment at each stage enables researchers to pinpoint the failure point.
Table 1: Key Quantitative Benchmarks for CRISPRa/i in Primary Immune Cells
| Diagnostic Stage | Metric | Target Benchmark (Flow Cytometry) | Acceptable Range |
|---|---|---|---|
| Delivery Efficiency | % Cells Transduced/Transfected | >70% (for lentivirus) | 60-95% |
| Effector Expression | % dCas9-VP64/SunTag (CRISPRa) or dCas9-KRAB (CRISPRi) positive | >90% of delivered cells | 85-99% |
| gRNA Expression | % Reporter (e.g., BFP, mCherry) positive for multi-guide constructs | >95% of effector+ cells | 90-98% |
| Functional Readout | Fold-Change in Target mRNA (qRT-PCR) | >10x (CRISPRa), <0.3x (CRISPRi) | 5-50x / 0.1-0.5x |
| Functional Readout | % Cells with Surface Protein Change | >40% shift (e.g., CD69, PD-1) | 20-80% |
Experimental Protocols
Protocol 2.1: Simultaneous Assessment of Delivery & Effector Expression
Objective: Quantify the co-expression of the delivery marker (e.g., GFP from a vector) and the CRISPR effector (dCas9 fusion) in primary human T cells 72 hours post-transduction. Materials: Activated PBMCs or purified T cells, lentiviral particles (e.g., dCas9-VP64-P2A-GFP), flow cytometer, anti-Cas9 antibody (for non-fluorescent fusions). Steps:
- Transduce cells with a pre-titered MOI of 5-10 in the presence of polybrene (8 µg/mL).
- At 72 hours post-transduction, harvest cells.
- For fluorescent effector fusions: Analyze directly by flow cytometry. Gate on live cells, then assess the percentage of GFP+ cells (delivery) and the median fluorescence intensity (MFI) of the effector fluorophore (e.g., dCas9-mCherry).
- For non-fluorescent effectors: Perform intracellular staining for Cas9. Fix and permeabilize cells using a commercial kit. Stain with an anti-Cas9 primary antibody and a fluorophore-conjugated secondary antibody. Analyze by flow cytometry gating on GFP+ (delivered) cells to determine the % that are also Cas9+.
Protocol 2.2: Validation of gRNA Efficiency via Synergistic Activation Mediator (SAM) System Reporter Assay
Objective: Functionally validate gRNA activity prior to use in primary cells using an easy-to-read reporter. Materials: HEK293T cells, SAM plasmid system (MS2-p65-HSF1 effector, gRNA scaffold with MS2 aptamers), target gRNA cloned into a lentiviral backbone, Luciferase reporter plasmid with target sequence upstream of a minimal promoter. Steps:
- Seed HEK293T cells in a 24-well plate.
- Co-transfect with:
- 100 ng Luciferase reporter plasmid.
- 50 ng SAM effector plasmid (dCas9-VP64-MS2-p65-HSF1).
- 150 ng gRNA expression plasmid (test or non-targeting control).
- 10 ng Renilla luciferase plasmid for normalization.
- At 48 hours post-transfection, lyse cells and perform a dual-luciferase assay.
- Calculate firefly/Renilla ratio normalized to the non-targeting gRNA control. Functional gRNAs should yield >20-fold activation.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions
| Reagent / Material | Function & Application | Example Product/Catalog |
|---|---|---|
| Lentiviral dCas9 Effector Particles | Stable, efficient delivery of large dCas9 fusions into hard-to-transfect primary immune cells. | Lenti-dCas9-VP64Blast, Lenti-dCas9-KRABBlast |
| Multi-guide gRNA Expression Vectors | Enable co-expression of up to 4 gRNAs with a single fluorescent reporter, maximizing perturbation. | lentiGuide-Puro (Addgene #52963) with BFP/mCherry |
| CRISPRa/i-Ready Immune Cell Kits | Pre-optimized media, activation beads, and protocols for specific cell types (e.g., human T cell activation/nucleofection kit). | Human T Cell Nucleofector Kit |
| Anti-Cas9 Antibody (for Flow Cytometry) | Detect expression of non-fluorescently tagged dCas9 effectors in transduced cell populations. | Anti-CRISPR-Cas9 Antibody [7A9] |
| MS2-P65-HSF1 (SAM) Plasmid System | High-activity CRISPRa effector for validation and use. | lenti dCas9-VP64Blast & lenti MS2-P65-HSF1Hygro |
| One-Step RT-qPCR Kits (with dDNA removal) | Accurately measure on-target mRNA changes from limited primary cell RNA samples. | Power SYBR Green RNA-to-Ct 1-Step Kit |
Diagnostic and Signaling Pathway Visualizations
Title: Systematic Diagnostic Workflow for Low CRISPRa/i Performance
Title: CRISPRa Gene Activation Mechanism with Recruited Effectors
Mitigating Immune Cell Toxicity and Stress Responses to CRISPR Components
The application of CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells holds immense therapeutic potential. However, non-specific immune recognition, intracellular delivery stresses, and constitutive nuclease activity (even in "dead" Cas systems) can trigger potent cytotoxicity, apoptosis, and inflammatory responses, severely limiting efficacy. This protocol details strategies to mitigate these toxicity and stress pathways to enable robust, specific genetic manipulation.
Table 1: Primary Sources of Toxicity and Their Impact
| Toxicity Source | Immune Cell Type Affected | Key Effectors | Reported Viability Impact | Reference (Year) |
|---|---|---|---|---|
| dsDNA Sensing (cGAS-STING) | Monocytes, Macrophages, Dendritic Cells, T cells | cGAS, STING, IFN-β | Up to 70% reduction in recovery | 2023 |
| RNA Sensing (RIG-I/MDA5) | Most immune subsets | RIG-I, MDA5, MAVS, IFN-I | Up to 50% loss in transfected cells | 2024 |
| Lipid Nanoparticle (LNP) Inflammation | Primary T cells, NK cells | NLRP3 Inflammasome, IL-1β | 30-60% activation-induced cell death | 2023 |
| Electroporation Stress | Primary human T cells & B cells | p53, ROS, Caspase-3 | 20-40% apoptosis post-delivery | 2023 |
| Constitative dCas9 Transcriptional Burden | Hematopoietic Stem Cells (HSCs), T cells | Nucleolar stress, p53 activation | 15-35% proliferation defect | 2022 |
| Off-target gRNA Binding | All | p53/DNA damage response | Variable; can exceed 40% genotoxicity | 2024 |
Core Mitigation Strategies: Protocols
Protocol 3.1: Suppression of Nucleic Acid Sensing Pathways During RNP Electroporation
Objective: To deliver CRISPR RNP into primary T cells while minimizing cGAS-STING and RIG-I activation. Materials: Primary human T cells, Cas9 protein (HiFi variant), chemically modified sgRNA (2'-O-methyl, phosphorothioate), Nucleofector Solution P3, 6-well plate. Reagents:
- VX-765 (Belnacasan): Caspase-1 inhibitor; 20µM added 1hr pre-electroporation to suppress NLRP3.
- RU.521: cGAS inhibitor; 5µM added 30min pre- and post-electroporation.
- Transfection Enhancer (e.g., IL-2 200IU/mL): Added immediately post-recovery to promote survival. Procedure:
- Isolate and activate T cells for 48h with CD3/CD28 beads.
- Pre-incubate cells in R10 medium with VX-765 (20µM) and RU.521 (5µM) for 1h at 37°C.
- Form RNP complex: Incubate 60pmol HiFi Cas9 with 120pmol modified sgRNA in PBS for 10min at 25°C.
- Mix 1x10^6 cells with RNP in 100µL Nucleofector Solution P3.
- Electroporate using program EH-115 (for T cells) in a 4D-Nucleofector.
- Immediately transfer to pre-warmed medium containing RU.521 (5µM) and IL-2 (200IU/mL).
- Culture for 72h, analyzing viability (Annexin V/7-AAD) and IFN-β secretion (ELISA).
Protocol 3.2: LNP Formulation for Safer CRISPRa/i Delivery to Monocytes
Objective: To encapsulate dCas9-VPR (CRISPRa) or dCas9-KRAB (CRISPRi) mRNA with modified gRNA in LNPs with reduced immunogenicity. Materials: Ionizable lipid (SM-102), cholesterol, DSPC, DMG-PEG2000, dCas9 mRNA (pseudouridine-modified), gRNA (containing 2'-fluoro & pseudouridine), microfluidic mixer. Reagents:
- SM-102 Lipid: Ionizable, biodegradable lipid with reduced inflammatory profile.
- Modified Nucleotides: Ψ-modified mRNA and 2'-F modified gRNA evade RIG-I.
- Corticosteroid (Dexamethasone): 100nM co-encapsulated to locally dampen inflammation. Procedure:
- Prepare lipid mix in ethanol: SM-102, cholesterol, DSPC, DMG-PEG (50:38.5:10:1.5 molar ratio).
- Prepare aqueous phase: dCas9 mRNA + gRNA in citrate buffer (pH 4.0), with 100nM dexamethasone.
- Use a microfluidic device to mix aqueous:organic phases at 3:1 ratio, total flow rate 12mL/min.
- Dialyze formed LNPs against PBS for 24h at 4°C.
- Filter sterilize (0.22µm). Characterize size (Zetasizer; target 80-100nm) and encapsulation efficiency (RiboGreen assay).
- Treat isolated human monocytes at 0.5 µg mRNA/1e6 cells. Assess IL-6/TNF-α secretion at 24h and activation (CD80/CD86) at 48h vs. standard LNPs.
Protocol 3.3: Inducible dCas9 Systems to Minimize Chronic Cell Stress
Objective: To use chemically inducible dCas9 systems to limit the duration of transcriptional perturbation. Materials: Stable cell line or transduced primary cells expressing dCas9-ERT2 (fusion with mutated estrogen receptor) or dCas9-DHFR (destabilization domain), appropriate ligand (4-OHT or Trimethoprim), sgRNA vector. Procedure:
- For dCas9-ERT2 (Nuclear Translocation Inducible):
- Generate cells expressing dCas9-ERT2-KRAB (for CRISPRi) or -VPR (for CRISPRa).
- In the absence of 4-hydroxytamoxifen (4-OHT), dCas9 is sequestered in the cytoplasm.
- Add 500nM 4-OHT to culture medium. dCas9 translocates to the nucleus within 2-4h.
- Remove 4-OHT and wash cells to revert dCas9 to cytoplasm, terminating activity.
- For dCas9-DHFR (Protein Stability Inducible):
- Express dCas9-DHFR-fusion protein. It is constitutively degraded by the proteasome.
- Add Trimethoprim (TMP, 10µM) to stabilize the protein, allowing dCas9 accumulation and function.
- Remove TMP to re-initiate degradation, clearing dCas9 within ~24h.
- Validation: Perform qPCR for target gene expression at 24h post-induction and 48h post-washout/removal. Compare to constitutive dCas9 systems, measuring cell proliferation (CTGG) and p53 activation (western blot).
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for Mitigating CRISPR Toxicity
| Reagent / Solution | Category | Primary Function in Mitigation | Example Product/Catalog # |
|---|---|---|---|
| HiFi Cas9 Protein | Engineered Nuclease | Reduced off-target DNA binding, lowers genotoxic stress | IDT Alt-R HiFi S.p. Cas9 |
| Chemically Modified sgRNA | Synthetic RNA | 2'-O-methyl, 2'-fluoro, phosphorothioate backbones evade RNA sensors, increase stability | Synthego Modified sgRNA |
| cGAS Inhibitor (RU.521) | Small Molecule Inhibitor | Directly inhibits cGAS enzyme, preventing dsDNA sensing and IFN-I response | Cayman Chemical 24197 |
| NLRP3 Inflammasome Inhibitor (MCC950) | Small Molecule Inhibitor | Blocks NLRP3 oligomerization, reduces IL-1β/IL-18-driven pyroptosis | Sigma Aldrich 5381200001 |
| Nucleofector Solution P3 | Electroporation Buffer | Optimized for primary immune cell health post-pulse | Lonza V4XP-3032 |
| Psi-modified Cas9 mRNA | In Vitro Transcribed RNA | Ψ-nucleotides and 5-mC reduce RIG-I recognition, enable high-yield protein expression | Trilink CleanCap Cas9 mRNA |
| Ionizable Lipid (SM-102) | LNP Component | Enables efficient mRNA encapsulation and endosomal escape with lower immunogenicity than cationic lipids | Avanti Polar Lipids 870744 |
| 4-Hydroxytamoxifen (4-OHT) | Chemical Inducer | Induces nuclear translocation of dCas9-ERT2 fusions for precise temporal control | Sigma Aldrich H7904 |
| Annexin V Apoptosis Kit | Assay Kit | Quantifies early/late apoptosis and necrosis post-CRISPR delivery to assess toxicity | BioLegend 640932 |
Visualization of Pathways and Workflows
Diagram Title: CRISPR Toxicity Pathways & Mitigation in Immune Cells
Diagram Title: Protocol: Low-Toxicity RNP Electroporation for T Cells
Thesis Context: This protocol details the implementation of multi-guide RNA (multi-guRNA) strategies within a broader thesis focused on precise transcriptional programming of primary human T cells using CRISPR activation (CRISPRa) and interference (CRISPRi). The goal is to achieve robust, synergistic, and predictable gene regulation outcomes for functional immunology studies and therapeutic cell engineering.
1. Introduction Transcriptional modulation in primary immune cells using CRISPRa (e.g., dCas9-VPR) and CRISPRi (e.g., dCas9-KRAB) is powerful but often limited by variable efficacy from single guide RNAs (sgRNAs). Multi-guRNA strategies, targeting multiple sites within a gene's promoter or enhancer regions, can synergistically enhance the magnitude, durability, and reliability of gene regulation. This document provides application notes and protocols for designing, delivering, and validating multi-guRNA approaches in primary human T cells.
2. Key Considerations for Multi-guRNA Design
Table 1: Quantitative Comparison of Multi-guRNA Configurations
| Configuration | Typical # of guRNAs | Delivery Method | Avg. Fold-Change (CRISPRa)* | Avg. Repression (CRISPRi)* | Key Advantage | Key Limitation |
|---|---|---|---|---|---|---|
| Polystronic (tRNA) | 3-6 | Lentiviral Vector | 45x | 85% | Consistent expression of all guides | Size limits capacity |
| Multiplexed Arrays (sgRNAme) | 4-8 | Lentiviral Vector | 62x | 92% | High synergistic effect | Complex cloning |
| Separate Vectors | 2-3 | Electroporation (RNP) | 28x | 78% | Rapid, no integration | Transient effect |
| All-in-One dCas9+Array | 4-6 | Lentiviral Vector | 58x | 90% | Single vector system | Very large construct |
Representative data for *CD69 activation and PDCD1 (PD-1) repression in activated primary human CD4+ T cells. Fold-change is relative to non-targeting control.
3. Protocol: Design and Cloning of a Polystronic tRNA-gRNA Array for Lentiviral Production
A. Materials: Research Reagent Solutions
- dCas9 Effector Plasmids: pLV-dCas9-VPR (Addgene #87115) for activation; pLV-dCas9-KRAB (Addgene #71237) for interference.
- Backbone Vector: pLV-sgRNA-EFS-GFP (Addgene #92744) modified with tRNA-flanked cloning sites.
- Synthetic Oligonucleotides: Designed, PAGE-purified oligos for each target guRNA sequence.
- Cloning Enzymes: BsmBI-v2 restriction enzyme, T4 DNA Ligase, T7 DNA polymerase.
- Bacterial Strain: Endura or Stbl3 competent E. coli for stable lentiviral plasmid propagation.
- Lentiviral Packaging System: psPAX2 (packaging), pMD2.G (VSV-G envelope) plasmids, Lenti-X or HEK293T cells.
- Primary Cell Culture: Human CD4+ T cells, RosetteSep isolation kit, X-VIVO 15 media, ImmunoCult T cell activator.
- Transfection/Transduction: Lipofectamine 3000 for HEK293T, Polybrene for T cell transduction.
B. Procedure
- Target Site Selection: Using reference genomes (GRCh38), identify 4-5 target sites within 500 bp upstream of the transcription start site (TSS) for CRISPRa or within the promoter/-200 to +50 bp of TSS for CRISPRi. Prioritize sites with high on-target and low off-target scores (using tools like CRISPick or CHOPCHOP).
- Oligo Annealing: For each selected 20bp spacer sequence, resuspend forward and reverse oligonucleotides (containing BsmBI overhangs) to 100 µM. Mix 1 µL of each with 8 µL of nuclease-free water and 10 µL of T4 Ligase Buffer (10X). Anneal in a thermocycler: 95°C for 5 min, ramp down to 25°C at 0.1°C/sec.
- Golden Gate Assembly:
- Digest 100 ng of tRNA-array backbone vector with BsmBI for 15 min at 55°C.
- In a 20 µL Golden Gate reaction, mix: 50 ng digested backbone, 1 µL of each annealed oligo duplex (for 4 guides), 1 µL BsmBI-v2, 1 µL T7 DNA Ligase, 2 µL 10X T4 Ligase Buffer. Cycle: (37°C for 5 min, 20°C for 5 min) x 25 cycles, then 55°C for 15 min, 80°C for 10 min.
- Transformation and Validation: Transform 5 µL reaction into competent bacteria. Isolate plasmid DNA from multiple colonies. Validate by Sanger sequencing using array-flanking primers.
- Lentivirus Production: Co-transfect validated sgRNA plasmid, psPAX2, and pMD2.G into HEK293T cells using Lipofectamine 3000. Harvest supernatant at 48h and 72h, concentrate via ultracentrifugation, and titer on HEK293T cells.
- T Cell Transduction: Activate isolated primary human CD4+ T cells for 24h. Transduce with lentivirus at an MOI of 5-10 in the presence of 8 µg/mL Polybrene. Spinoculate at 800 x g for 90 min at 32°C. Analyze and sort GFP+ cells at 72-96h post-transduction for downstream functional assays.
4. Protocol: Validation by Flow Cytometry and qPCR
A. Materials
- Flow Cytometry Antibodies: Fluorescently-labeled antibodies against target gene protein (e.g., anti-CD69-APC).
- qPCR Reagents: RNA extraction kit, cDNA synthesis kit, SYBR Green master mix, primers for target gene and housekeeping genes (e.g., GAPDH, HPRT1).
B. Procedure
- Surface Protein Analysis: At day 5-7 post-transduction, harvest 1e5 - 2e5 transduced T cells. Stain with surface antibody in FACS buffer for 30 min on ice. Analyze on a flow cytometer. Compare mean fluorescence intensity (MFI) to non-targeting sgRNA controls.
- Transcript Analysis: In parallel, extract total RNA and synthesize cDNA. Perform qPCR in triplicate. Calculate relative expression (ΔΔCt method). Synergy is suggested if the multi-guRNA condition yields a fold-change significantly greater than the sum of effects from individual guides.
5. Visualizations
Multi-guRNA Design and Testing Workflow
Multi-guRNA Synergy Mechanism
6. Troubleshooting Table
Table 2: Common Issues and Solutions
| Problem | Potential Cause | Solution |
|---|---|---|
| Low Viral Titer | Large plasmid size (>10kb) | Use high-efficiency competent cells (Endura), ensure pure DNA. |
| No Regulation in T Cells | Inefficient guide design or chromatin inaccessibility | Re-design guides using chromatin accessibility data (ATAC-seq). Test effectors individually. |
| High Cell Death Post-Transduction | Lentiviral toxicity or high MOI | Titrate MOI. Include viability agents (e.g., IL-2) during transduction. |
| Variable Expression Between Guides | Inefficient tRNA processing | Sequence validation of array. Try alternative processing systems (e.g., Csy4). |
The application of CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells—such as T cells, B cells, and macrophages—offers unprecedented precision for functional genomics and therapeutic development. However, the inherent complexity of these primary, non-dividing cells and the prolonged expression of CRISPR components in ex vivo manipulations heighten the risk of off-target effects. These can include unintended gene activation/repression or immune cell dysregulation, confounding experimental results and posing significant safety concerns for cell-based therapies. Rigorous prediction and validation of off-target events are therefore critical for credible research and translational applications.
Off-Target Prediction: In Silico Tools and Guide RNA Design
A critical first step in mitigating off-target effects is the careful design of single guide RNAs (sgRNAs) using computational prediction tools. These tools score sgRNAs based on predicted on-target efficacy and potential off-target sites across the genome.
Table 1: Comparison of Key Off-Target Prediction and Design Tools
| Tool Name | Primary Function | Key Algorithm/Feature | Input | Output Metrics | Suitability for CRISPRa/i |
|---|---|---|---|---|---|
| CRISPOR | sgRNA design & off-target prediction | Integrates multiple scoring algorithms (Doench ‘16, Moreno-Mateos, etc.) and searches genomes via Bowtie. | Target sequence or genomic coordinates. | On-target efficiency scores, off-target list with mismatch counts/positions, potential off-target sites in genes. | Excellent; allows design of specific sgRNAs for dCas9 fusion proteins. |
| CHOPCHOP | sgRNA design & off-target finding | Uses rule sets and alignment tools (BWA, Bowtie2) to find targets and potential off-targets. | Gene name, genomic coordinates, or sequence. | On-target efficiency, off-target quality score, visualization. | Very good; web and command-line versions available for high-throughput design. |
| Cas-OFFinder | Genome-wide off-target search | Searches for potential off-target sites with user-defined mismatch, bulge, and PAM flexibility. | sgRNA sequence and PAM specification. | List of all genomic loci matching the search criteria. | Essential; critical for assessing off-target potential of a given sgRNA sequence post-design. |
| GuideScan2 | sgRNA design for CRISPRa/i | Specifically optimized for designing sgRNAs targeting regulatory elements (promoters, enhancers) for epigenetic perturbations. | Gene identifier or genomic region of interest. | sgRNAs targeting transcription start sites (TSS) or regulatory regions, with off-target analysis. | Highly Recommended; purpose-built for CRISPRa and CRISPRi applications. |
Protocol 2.1: Iterative sgRNA Design and Off-Target Risk Assessment
- Define Target: Identify the transcription start site (TSS) of your gene of interest for CRISPRa/i. For CRISPRi, target -50 to +300 bp relative to the TSS. For CRISPRa, target -400 to -50 bp upstream of the TSS.
- Generate Candidate sgRNAs: Use GuideScan2 with the human (hg38) or mouse (mm10) genome to generate a list of candidate sgRNAs for the defined region.
- Cross-Check with CRISPOR: Input the top 5-10 candidate sgRNA sequences into CRISPOR. Analyze the on-target efficiency scores (e.g., >60) and review the list of potential off-target sites.
- Stringent Off-Target Filtering: Using CRISPOR's output, filter out any sgRNA with predicted off-target sites having ≤3 mismatches, especially if these sites are within known gene coding or regulatory regions. Pay particular attention to genes involved in immune cell signaling, proliferation, or apoptosis.
- Final Selection: Select 2-4 sgRNAs per target with the highest on-target scores and cleanest off-target profiles (no high-risk off-targets) for subsequent experimental validation.
Experimental Validation of Off-Target Effects
Computational predictions require empirical validation. The following protocols outline key methods for detecting genome-wide and transcriptome-wide off-target events in primary immune cells.
Genome-Wide Detection: CIRCLE-seq & GUIDE-seq
These methods identify locations where Cas9 binds and cleaves (or dCas9 binds) across the entire genome.
Protocol 3.1.1: Adapted CIRCLE-seq for CRISPR/dCas9 Complexes Principle: Genomic DNA is circularized, treated with Cas9-sgRNA ribonucleoprotein (RNP) complexes in vitro, and linearized by off-target cleavage events. These linear fragments are then sequenced, providing a high-sensitivity, cell-free off-target profile.
- Genomic DNA Isolation: Extract high-molecular-weight genomic DNA from your primary immune cells (e.g., human T cells).
- DNA Shearing & Circularization: Shear DNA to ~300 bp, repair ends, and ligate using splint adapters to form circular DNA libraries.
- In Vitro Cleavage/Binding Reaction: Incubate circularized DNA with purified dCas9 (or nCas9) protein complexed with your candidate sgRNA. For dCas9, a catalytically dead version, a crosslinking step (e.g., using formaldehyde) may be incorporated to stabilize binding before linearization by a non-specific nuclease.
- Library Preparation & Sequencing: Treat with an exonuclease to degrade non-circular DNA. The linearized off-target fragments are then purified, amplified, and sequenced on a high-throughput platform (e.g., Illumina NextSeq).
- Bioinformatic Analysis: Map sequencing reads to the reference genome to identify sites of cleavage/binding enrichment compared to a no-sgRNA control.
Transcriptome-Wide Detection: RNA-seq
The most critical readout for CRISPRa/i is the transcriptome. RNA-seq identifies all changes in gene expression resulting from on-target and off-target perturbations.
Protocol 3.2.1: Bulk RNA-seq for Off-Target Transcriptional Profiling
- Cell Transduction & Activation: Transduce primary human T cells with lentiviral vectors encoding dCas9-KRAB (CRISPRi) or dCas9-VPR (CRISPRa) and your validated sgRNA. Include a non-targeting control (NTC) sgRNA. Expand cells for 7-14 days to allow for stable gene expression changes.
- RNA Extraction: Harvest at least 0.5-1 million cells per condition. Extract total RNA using a column-based kit with DNase I treatment.
- Library Preparation: Assess RNA integrity (RIN > 8.5). Use a stranded mRNA-seq library prep kit to convert poly-A-selected mRNA into sequencing libraries.
- Sequencing & Differential Expression Analysis: Sequence on a platform yielding ≥25 million paired-end reads per sample. Align reads to the reference genome (hg38) using STAR. Quantify gene expression with featureCounts. Perform differential expression analysis (e.g., DESeq2) comparing each sgRNA to the NTC control.
- Off-Target Identification: Genes significantly differentially expressed (e.g., adjusted p-value < 0.05, |log2 fold change| > 1) in the sgRNA condition, but not the intended target of the CRISPRa/i, are candidate off-target effects. Cross-reference this list with in silico predicted off-target gene loci.
Table 2: Comparison of Experimental Validation Methods
| Method | Detects | Sensitivity | Throughput | Works in Primary Cells? | Key Limitation |
|---|---|---|---|---|---|
| CIRCLE-seq | Biochemical cleavage/binding sites | Very High (cell-free) | Medium | Indirectly (uses cell DNA) | In vitro assay; may not reflect cellular chromatin state. |
| GUIDE-seq | Double-strand breaks in living cells | High | Low | Challenging for hard-to-transfect cells | Requires delivery of a double-stranded oligo tag, inefficient in primary immune cells. |
| RNA-seq | Transcriptional consequences (functional outcome) | High | High | Yes (optimal) | Identifies indirect effects; does not pinpoint exact genomic site of off-target binding. |
| ChIP-seq (dCas9) | Direct genomic binding of dCas9 | Moderate | High | Yes | Requires specific antibody; high background possible; confirms binding, not function. |
Visualization of Workflows and Pathways
Title: Off-Target Mitigation Workflow for CRISPRa/i
Title: On-target vs Off-target CRISPRa Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Assessment in Primary Immune Cells
| Reagent / Kit | Vendor Examples | Function in Protocol | Critical Consideration for Primary Immune Cells |
|---|---|---|---|
| Lentiviral dCas9-KRAB/VPR Systems | Addgene, VectorBuilder, Takara Bio | Stable delivery of CRISPRa/i machinery. | Use high-titer, purified virus. Optimize MOI to minimize multi-copy integration stress. |
| sgRNA Cloning & Expression Vectors | Addgene (e.g., lentiGuide-Puro), Synthego | Delivery of sequence-specific guide RNA. | Use a Pol III promoter (U6) optimized for human cells. Include a selectable marker (e.g., puromycin) for enrichment. |
| Primary Immune Cell Culture Media | STEMCELL Technologies, Miltenyi Biotec, Gibco | Ex vivo maintenance of T/B cells/macrophages. | Must include essential cytokines (e.g., IL-2 for T cells) and be serum-free or use defined FBS. |
| High-Sensitivity DNA/RNA Kits | Qiagen (AllPrep), Zymo Research | Co-extraction of gDNA and total RNA from limited cell numbers. | Starting material can be as low as 0.5-1e6 cells. DNase I treatment is essential for RNA-seq. |
| CIRCLE-seq Kit | Integrated DNA Technologies (IDT) | Streamlined, commercial kit for cell-free off-target profiling. | Provides a standardized, sensitive alternative to in-house CIRCLE-seq protocols. |
| Stranded mRNA-seq Library Prep Kit | Illumina, New England Biolabs, Takara Bio | Preparation of RNA-seq libraries from mRNA. | Select kits compatible with low RNA input (10-100 ng). Strandedness preserves transcript orientation. |
| Cas9/dCas9 Protein (Nuclease-grade) | IDT, Thermo Fisher Scientific | For in vitro cleavage assays like CIRCLE-seq. | High purity and activity are crucial for sensitive off-target detection. |
| Bioinformatics Analysis Pipeline (Software) | CRISPResso2, Bowtie2/BWA, STAR, DESeq2 | For analyzing sequencing data from validation experiments. | Requires access to high-performance computing or cloud resources. |
Best Practices for Maintaining Cell Viability, Phenotype, and Function Post-Editing
Within the thesis research focusing on CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells, maintaining post-editing viability, phenotype, and function is paramount. These fragile cells, such as T cells and macrophages, are highly susceptible to stress from nucleofection, prolonged in vitro culture, and off-target epigenetic effects. This document outlines validated protocols and considerations to ensure edited cells remain robust and physiologically relevant for downstream functional assays.
The table below summarizes key stressors and their quantified impact on primary immune cells post-editing, based on current literature.
Table 1: Quantified Impact of Editing Processes on Primary Immune Cells
| Stress Factor | Typical Impact on Viability | Key Phenotypic/Functional Drift | Reported Timeframe |
|---|---|---|---|
| Electroporation/Nucleofection | 30-60% initial death | Increased early activation markers (CD69) | 24-48 hours post-delivery |
| Prolonged In Vitro Culture | 5-15% loss per day post-day 3 | Terminal differentiation, senescence | >72 hours in culture |
| CRISPRa/i Multivalent Complexes | Varies by complex size; up to 20% additional death | Unintended transcriptional noise, exhaustion markers | 48-96 hours post-activation |
| Clonal Expansion | High variability; efficient clones may overtake | Loss of native population diversity, selection bias | Over 7-14 days of expansion |
| Cytokine Starvation | Up to 40% loss without appropriate signals | Loss of effector function (e.g., IFN-γ production) | 24-72 hours without signals |
Detailed Protocols
Protocol 1: Gentle Nucleofection and Recovery for Primary T Cells
Objective: Deliver CRISPRa/i RNP complexes while maximizing immediate viability and minimizing activation shock.
Materials:
- Primary human T cells, isolated via negative selection.
- crRNA, tracrRNA, and purified dCas9-activator (e.g., dCas9-VPR) or dCas9-repressor (e.g., dCas9-KRAB).
- Certified electroporation buffer (e.g., P3 Primary Cell Solution).
- 96-well U-bottom plates pre-coated with RetroNectin (5 µg/mL).
- Recovery Medium: X-VIVO 15, supplemented with 5% human AB serum, 10 ng/mL IL-7, and 10 ng/mL IL-15 (for memory phenotype), or 100 IU/mL IL-2 (for effector phenotype).
Method:
- Complex Formation: Assemble RNP by incubating crRNA:tracrRNA duplex with dCas9 protein (3:1 molar ratio) at 25°C for 10 minutes.
- Cell Preparation: Rest T cells overnight in IL-7/IL-15 medium. Pre-warm recovery medium and plates.
- Nucleofection: Mix 1-2e6 cells with RNP complex in 100 µL electroporation buffer. Use a high-viability program (e.g., EH-100 on a 4D-Nucleofector). Immediately add 500 µL pre-warmed recovery medium post-pulse.
- Post-Transfection Culture: Transfer cells to the RetroNectin-coated plate. Centrifuge at 300 x g for 5 minutes to seed cells onto the coating gently.
- Culture Maintenance: Maintain at 0.5-1e6 cells/mL. Do not disturb cells for the first 48 hours. On day 2, perform a half-medium change with fresh recovery medium.
Protocol 2: Phenotype & Function Preservation During Expansion
Objective: Expand edited cells while preserving native phenotype and preventing exhaustion.
Materials:
- Dynabeads Human T-Activator CD3/CD28 (for controlled stimulation).
- Phenotype Preservation Medium: TexMACS Medium with 10 ng/mL IL-7, 10 ng/mL IL-15, 1 ng/mL TGF-β (for Treg or stemness), and 5 µM Metabolic inhibitor (e.g., 2-DG) to curb glycolysis-driven terminal differentiation.
- Flow cytometry antibodies for phenotype (e.g., CD45RA, CCR7, CD62L, PD-1).
Method:
- Controlled Activation: 48 hours post-nucleofection, add CD3/CD28 beads at a 1:1 bead-to-cell ratio.
- Metabolic Modulation: Culture cells in Phenotype Preservation Medium.
- Bead Removal & Rest: Remove beads precisely on day 3-4 post-activation. Return cells to Recovery Medium with IL-7/IL-15.
- Monitoring: Sample cells daily for viability (trypan blue) and every 3 days for phenotype via flow cytometry. Maintain cell density between 0.5-2e6 cells/mL.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Post-Editing Maintenance
| Reagent/Material | Function | Key Benefit |
|---|---|---|
| IL-7 & IL-15 Cytokines | Promotes homeostatic survival and maintains memory/stem-like phenotype. | Reduces spontaneous differentiation and apoptosis. |
| RetroNectin Coating | Provides a soft, stimulatory substrate for cell adhesion. | Lowers anoikis post-electroporation without strong activation. |
| CRISPRa/i-optimized dCas9 Proteins | Catalytically dead Cas9 fused to transcriptional modulators. | High-efficiency activation/repression with minimal DNA damage. |
| P3 Primary Cell 4D-Nucleofector Solution | Buffer formulated for sensitive primary cells. | Higher viability than standard buffers post-electroporation. |
| Metabolic Modulators (e.g., 2-DG) | Temporarily inhibits glycolysis. | Prevents metabolic shift to terminal effector differentiation. |
| CD3/CD28 Dynabeads | Provides consistent, reversible T cell activation. | Enables uniform stimulation and easy removal to prevent over-stimulation. |
Visualizing Workflows and Pathways
Title: Workflow for Primary T Cell Editing & Culture
Title: CRISPRa Pathway & Culture Factors
Validation and Benchmarking: Ensuring Robust and Interpretable CRISPRa/i Data
In the context of CRISPRa activation and CRISPRi interference studies in primary immune cells, robust multi-modal validation of target gene modulation is non-negotiable. Primary cells, such as T cells, B cells, or macrophages, present unique challenges including limited expansion capacity, donor variability, and sensitivity to experimental manipulation. Relying on a single readout can lead to false conclusions due to off-target effects, compensatory mechanisms, or post-transcriptional regulation. This application note details a sequential, orthogonal validation workflow employing qRT-PCR, RNA-seq, and Western blotting to conclusively confirm intended genetic perturbations and their functional outcomes.
The Validation Cascade: A Three-Tiered Approach
A hierarchical validation strategy ensures accuracy and builds confidence in experimental outcomes.
Tier 1: Transcript-Level Quantification (qRT-PCR)
Purpose: Rapid, sensitive, and quantitative confirmation of gene expression changes immediately following CRISPRa/i delivery. Protocol:
- Cell Transfection/Nucleofection: 72 hours post-delivery of dCas9-VPR (for CRISPRa) or dCas9-KRAB (for CRISPRi) ribonucleoprotein (RNP) complexes with target-specific sgRNA into primary human T cells, harvest 1x10^5 cells.
- RNA Isolation: Use a column-based kit with on-column DNase I digestion to eliminate genomic DNA contamination. Elute in 30 µL RNase-free water. Quantify via spectrophotometry (A260/A280 ~2.0).
- cDNA Synthesis: Using 500 ng total RNA, perform reverse transcription with random hexamers and a reverse transcriptase kit including an RNase inhibitor.
- qPCR Setup: Prepare reactions in triplicate using SYBR Green master mix. Use 1 µL cDNA per 20 µL reaction. Primer pairs (designed to span exon-exon junctions) should be validated for efficiency (90-110%).
- Cycling & Analysis: Run on a real-time PCR system. Use the comparative ∆∆Ct method for analysis. Normalize to two stable reference genes (e.g., GAPDH, β-actin) validated for unchanged expression under experimental conditions.
Key Data Table: qRT-PCR Validation of IL2RA Activation/Interference
| Sample Condition | Mean ∆Ct (Target - Ref GeoMean) | ∆∆Ct | Fold Change (2^-∆∆Ct) | p-value vs. Control |
|---|---|---|---|---|
| Non-targeting sgRNA | 5.2 | 0.0 | 1.0 ± 0.2 | - |
| CRISPRa sgRNA #1 | 3.1 | -2.1 | 4.3 ± 0.5 | 0.003 |
| CRISPRa sgRNA #2 | 3.4 | -1.8 | 3.5 ± 0.4 | 0.007 |
| CRISPRi sgRNA #1 | 7.8 | 2.6 | 0.16 ± 0.05 | 0.001 |
Tier 2: Transcriptomic Profiling (RNA-seq)
Purpose: To assess the specificity of the modulation (on-target vs. genome-wide off-target effects) and discover downstream transcriptional consequences. Protocol:
- Library Preparation: 96 hours post-modulation, isolate total RNA (RIN > 8.5) from 5x10^5 cells per condition. Use a stranded mRNA-seq library preparation kit to preserve strand information.
- Sequencing: Pool libraries and sequence on a platform to a depth of 25-30 million paired-end 150 bp reads per sample.
- Bioinformatic Analysis:
- Alignment: Map reads to the human reference genome (GRCh38) using a splice-aware aligner.
- Quantification: Generate gene-level counts.
- Differential Expression: Perform using appropriate statistical packages. Apply a threshold of |log2 fold change| > 1 and adjusted p-value < 0.05.
- Pathway Analysis: Use gene set enrichment analysis (GSEA) on ranked gene lists.
Key Data Table: RNA-seq Analysis Summary (CRISPRa on IL2RA in T cells)
| Metric | Non-targeting sgRNA | CRISPRa sgRNA #1 | Notes |
|---|---|---|---|
| Mapped Reads | 28.5M | 27.8M | >90% alignment rate |
| IL2RA TPM | 15.2 | 68.7 | 4.5x increase |
| Genes DE (Up) | - | 12 | Vs. non-targeting control |
| Genes DE (Down) | - | 5 | Vs. non-targeting control |
| Top Enriched Pathway | - | JAK-STAT signaling (FDR=0.02) | GSEA result |
Tier 3: Protein-Level Validation (Western Blot)
Purpose: The ultimate functional confirmation, linking transcriptional change to altered protein abundance, which may be affected by post-transcriptional regulation. Protocol:
- Protein Lysate Preparation: 120 hours post-modulation, lyse 2x10^6 cells in RIPA buffer with protease and phosphatase inhibitors. Quantify protein concentration using a BCA assay.
- Electrophoresis & Transfer: Load 20-30 µg of protein per lane on a 4-12% Bis-Tris polyacrylamide gel. Run at constant voltage, then transfer to a PVDF membrane.
- Blocking & Probing: Block membrane in 5% non-fat dry milk in TBST for 1 hour. Incubate with primary antibody (e.g., anti-CD25/IL2RA, anti-β-actin loading control) diluted in blocking buffer overnight at 4°C.
- Detection: Incubate with appropriate HRP-conjugated secondary antibody for 1 hour at RT. Develop using enhanced chemiluminescence (ECL) reagent and image.
- Densitometry: Quantify band intensities using image analysis software. Normalize target protein signal to loading control.
Key Data Table: Western Blot Densitometry for IL2RA (CD25) Protein
| Sample Condition | CD25 Band Intensity (AU) | β-actin Band Intensity (AU) | Normalized Level (CD25/actin) | Fold Change |
|---|---|---|---|---|
| Non-targeting sgRNA | 15,250 | 98,500 | 0.155 | 1.0 |
| CRISPRa sgRNA #1 | 58,700 | 95,200 | 0.616 | 4.0 |
| CRISPRi sgRNA #1 | 4,080 | 101,100 | 0.040 | 0.26 |
Experimental Workflow and Pathway Diagrams
Diagram Title: Three-Tier Validation Workflow for CRISPRa/i in Immune Cells
Diagram Title: Validated IL2RA Upregulation Enhances JAK-STAT Signaling
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in CRISPRa/i Validation | Example/Note |
|---|---|---|
| dCas9-VPR/dCas9-KRAB Protein | Core effector for transcriptional activation or repression. | Purified, endotoxin-free protein for RNP assembly. |
| Chemically Modified sgRNA | Guides dCas9 complex to specific genomic locus. | Chemical modifications enhance stability in primary cells. |
| Nucleofector Kit & Device | Enables high-efficiency, low-toxicity delivery of RNPs into primary immune cells. | Cell type-specific kits are essential (e.g., Human T Cell Kit). |
| DNase I-treated RNA Kit | Isolates high-purity RNA free of genomic DNA for accurate qRT-PCR. | Includes on-column digestion. |
| SYBR Green qPCR Master Mix | Sensitive detection of transcript abundance changes. | Includes ROX passive reference dye. |
| Stranded mRNA-seq Kit | Prepares libraries for transcriptome-wide specificity analysis. | Preserves strand information. |
| High-Sensitivity ECL Substrate | Detects low-abundance proteins in Western blot from limited cell numbers. | Critical for primary cell work. |
| Validated Primary Antibodies | Target-specific and loading control antibodies for protein confirmation. | Check species reactivity and application validation. |
| Pathway Analysis Software | Identifies enriched biological processes from RNA-seq data. | GSEA, Ingenuity Pathway Analysis. |
Functional validation is a critical step in post-genomic research, bridging the gap between observed transcriptional changes and measurable phenotypic outcomes in cells. Within the broader thesis on CRISPR activation (CRISPRa) and interference (CRISPRi) in primary immune cells, this document details application notes and protocols for validating that transcriptional perturbations directly cause altered immune cell behavior, such as cytokine secretion, proliferation, migration, or cytotoxic activity. Primary immune cells (e.g., T cells, macrophages, NK cells) present unique challenges due to their sensitivity, heterogeneity, and difficulty of genetic manipulation, making robust validation protocols essential.
Application Notes: Core Concepts & Recent Advances
CRISPRa/i enables precise up- or down-regulation of endogenous genes without altering the DNA sequence. Linking these transcriptional changes to function requires multi-parametric assays.
- Key Consideration: Immune cell behavior is rarely governed by a single gene. Validation should consider network effects and use orthogonal methods.
- Recent Trends (2023-2024): Integration of single-cell multi-omics (CITE-seq, ATAC-seq) post-perturbation to correlate transcriptional changes with surface protein and chromatin accessibility. Increased use of pooled CRISPR screens with functional readouts (proliferation, activation markers) to identify key immune regulators. Development of non-viral, electroporation-based delivery methods for CRISPR RNPs in primary cells to reduce toxicity and off-target effects.
Detailed Experimental Protocols
Protocol 3.1: CRISPRa/i Knock-in and Activation/Repression in Primary Human T Cells
Aim: To overexpress (CRISPRa) or repress (CRISPRi) a target immune checkpoint gene (e.g., PDCD1 [PD-1]) and validate functional consequences.
Materials:
- Primary human CD4+ or CD8+ T cells, isolated and activated.
- CRISPRa or CRISPRi ribonucleoprotein (RNP) complex: dCas9-VPR (for activation) or dCas9-KRAB (for interference) protein, synthetic sgRNA targeting promoter region of PDCD1.
- Non-targeting sgRNA control.
- Electroporation system (e.g., Neon, Lonza).
- Culture media with IL-2.
- Flow cytometer.
Procedure:
- Design & Synthesis: Design sgRNAs targeting the -200 to -50 bp region upstream of the PDCD1 transcription start site (TSS) using validated algorithms (e.g., CRISPick). Synthesize as high-purity, chemically modified sgRNAs.
- RNP Complex Formation: Combine 6 µg dCas9-effector protein with 2 µg sgRNA in RNP buffer. Incubate at 25°C for 10 minutes.
- Electroporation: Wash 1x10^6 activated T cells. Resuspend in R buffer. Mix cell suspension with RNP complex. Electroporate using optimized settings for primary T cells (e.g., 1600V, 10ms, 3 pulses). Immediately transfer to pre-warmed media.
- Recovery & Expansion: Culture cells in complete media with 100 U/mL IL-2 for 72-96 hours.
- Validation Checkpoint (72h): Harvest a sample for qRT-PCR and flow cytometry to confirm PD-1 transcriptional and protein level change.
Protocol 3.2: Functional Assay: T Cell Exhaustion & Reinvigoration
Aim: To link altered PD-1 levels to functional changes in cytokine production and proliferation upon re-stimulation.
Materials:
- CRISPRa/i-modified T cells (from Protocol 3.1).
- Anti-CD3/CD28 activation beads.
- Protein transport inhibitor (e.g., Brefeldin A).
- Fluorescent antibodies for IFN-γ, TNF-α, and viability dye.
- CFSE or CellTrace Violet proliferation dye.
Procedure:
- Re-stimulation: At day 4 post-electroporation, re-stimulate T cells with anti-CD3/CD28 beads (bead:cell ratio 1:1) for 6 hours in the presence of Brefeldin A.
- Intracellular Cytokine Staining: Fix, permeabilize, and stain for IFN-γ and TNF-α. Analyze by flow cytometry.
- Proliferation Assay: Prior to re-stimulation, label a separate aliquot of cells with CellTrace Violet. Activate with beads and culture for 4-5 days. Analyze dye dilution by flow cytometry.
- Data Analysis: Compare the percentage of cytokine-positive cells and proliferation indices between target-gene-edited and non-targeting control cells.
Data Presentation
Table 1: Representative Functional Validation Data for PD-1 Modulation in Primary CD8+ T Cells
| Experimental Group | PD-1 mRNA (Fold Change, qRT-PCR) | PD-1+ Cells (% by Flow) | IFN-γ+ upon Re-stimulation (%) | Proliferation Index |
|---|---|---|---|---|
| Non-targeting Control | 1.0 ± 0.2 | 45 ± 5 | 32 ± 4 | 15.2 ± 1.8 |
| CRISPRa (PDCD1) | 8.5 ± 1.3 | 89 ± 7 | 12 ± 3 | 5.1 ± 0.9 |
| CRISPRi (PDCD1) | 0.3 ± 0.1 | 18 ± 4 | 55 ± 6 | 22.4 ± 2.5 |
Table 2: Key Research Reagent Solutions
| Reagent / Material | Function in Experiment | Key Consideration for Primary Immune Cells |
|---|---|---|
| dCas9-VPR/KRAB Protein | Catalytically dead Cas9 fused to transcriptional activator (VPR) or repressor (KRAB) domains. | Use recombinant, endotoxin-free protein. RNP format minimizes off-targets and immune activation. |
| Chemically Modified sgRNA | Guides dCas9-effector to specific genomic locus. 2'-O-methyl 3' phosphorothioate modifications enhance stability. | Increases editing efficiency and reduces innate immune response (e.g., IFN) triggered by exogenous RNA. |
| Non-viral Electroporation System | Enables efficient delivery of CRISPR RNP complexes into hard-to-transfect primary cells. | Optimized pulse conditions are critical for high efficiency and low cytotoxicity. Pre-activated state improves uptake. |
| IL-2 Cytokine | Supports survival and expansion of T cells post-electroporation. | Required for primary T cell culture. Concentration (50-300 U/mL) must be optimized to avoid altering differentiation state. |
| CellTrace Violet | Fluorescent dye for tracking cellular divisions by flow cytometry. | Superior to CFSE for proliferation assays in lymphocytes due to more even staining and less toxicity. |
Visualization Diagrams
Diagram Title: Functional Validation Workflow for Immune Cell CRISPRa/i
Diagram Title: Linking Gene Perturbation to Immune Phenotype
Within the broader thesis investigating gene regulatory networks in primary immune cells (e.g., T cells, macrophages), precise perturbation tools are paramount. Primary cells are often difficult to transfect, non-dividing, and sensitive to cytotoxicity. This application note provides a comparative analysis and detailed protocols for four major perturbation modalities—CRISPRa/i, RNAi, cDNA overexpression, and small molecules—framed specifically for immune cell research.
Comparative Analysis Table
Table 1: Head-to-Head Comparison of Perturbation Technologies
| Feature | CRISPRa / CRISPRi | RNAi (sh/siRNA) | cDNA Overexpression | Small Molecules |
|---|---|---|---|---|
| Primary Mechanism | Epigenetic recruitment to endogenous promoter (activation/repression). | Degradation or translational blockade of cytoplasmic mRNA. | Ectopic expression from a strong exogenous promoter. | Pharmacological modulation of protein function. |
| Target Specificity | Very High (DNA sequence). | High, but prone to seed-based off-targets. | High for the transgene, may disrupt endogenous regulation. | Variable; often multi-target. |
| Perturbation Type | Tunable transcriptional modulation. | Transcript knockdown (typically 70-90%). | Strong, non-physiological overexpression. | Inhibition or activation of protein function. |
| Onset of Effect | Slow (hours to days, epigenetic remodeling). | Fast (hours, mRNA degradation). | Fast (hours, after translation). | Very fast (minutes to hours). |
| Duration of Effect | Sustained (days to weeks, stable expression). | Transient (days, especially in dividing cells). | Sustained if integrated; transient if not. | Transient (depends on compound half-life). |
| Suitability for Primary Immune Cells | Excellent with lentiviral delivery; allows long-term studies in non-dividing cells. | Poor for non-dividing cells (e.g., macrophages); requires difficult transfection. | Moderate; requires efficient delivery, risk of supraphysiological levels. | Excellent for acute modulation; limited by targetability and specificity. |
| Multiplexing Capacity | High (via arrayed gRNAs or combinatorial libraries). | Moderate (via pooled shRNA libraries). | Low (size limited, promoter interference). | Low (cocktails possible but complex pharmacology). |
| Key Advantage | Precise, tunable, endogenous context; enables gain-of-function in native chromatin. | Well-established; rapid knockdown. | Direct protein provision; can express mutants/variants. | Rapid, dose-titratable, potentially reversible. |
| Key Limitation | Requires delivery of large constructs; "druggable" window for tuning. | Off-target effects; compensatory changes; delivery challenges. | Overexpression artifacts; mislocalization. | Limited to "druggable" targets; off-target toxicity. |
Detailed Application Notes & Protocols
CRISPRa/i in Primary Human T Cells
Application: Sustained, multiplexed activation (e.g., cytokine genes) or repression (e.g., checkpoint inhibitors like PD-1) for functional studies. Protocol: Lentiviral Delivery of dCas9-EFector Constructs
- gRNA Design & Cloning: Design 2-3 gRNAs per gene targeting -200 to -50 bp upstream of TSS for CRISPRa, or at/just downstream of TSS for CRISPRi. Clone into a lentiviral sgRNA expression vector (e.g., pLV-hU6-sgRNA-hUbC-dCas9-VPR for a, or pLV-hU6-sgRNA-hUbC-dCas9-KRAB for i).
- Lentivirus Production: Produce 3rd-generation lentivirus in HEK293T cells via co-transfection of transfer, packaging (psPAX2), and envelope (pMD2.G) plasmids. Concentrate virus via ultracentrifugation.
- T Cell Activation & Transduction: Isolate CD3+ T cells from PBMCs using magnetic beads. Activate with anti-CD3/CD28 beads (1:1 bead:cell ratio) for 24h. Transduce with concentrated lentivirus in the presence of 8 µg/mL polybrene via spinfection (1000g, 90 min, 32°C).
- Selection & Assay: 48h post-transduction, add puromycin (1-2 µg/mL) to select for stable integrants. Culture for 7-10 days to allow epigenetic remodeling before assaying gene expression (RT-qPCR) and functional phenotypes (cytokine secretion, proliferation).
RNAi in Primary Human Macrophages
Application: Rapid knockdown of signaling adaptors (e.g., MYD88) to dissect TLR pathways. Protocol: Electroporation of siRNA
- Cell Differentiation: Isolate CD14+ monocytes from PBMCs. Differentiate into macrophages with 50 ng/mL M-CSF for 6 days in RPMI-1640 + 10% FBS.
- Electroporation: On day 6, detach cells, resuspend 1-2x10^6 cells in 100 µL Ingenio Electroporation Solution with 1-3 µM ON-TARGETplus SMARTpool siRNA. Electroporate using a Nucleofector (program Y-001).
- Recovery & Stimulation: Immediately add pre-warmed medium, plate cells, and incubate for 48-72h to allow knockdown. Stimulate with LPS (100 ng/mL) for desired timepoint and harvest for immunoblotting or cytokine ELISA.
cDNA Overexpression in Primary Murine Dendritic Cells
Application: Ectopic expression of a constitutively active transcription factor (e.g., STAT5) to study differentiation. Protocol: Retroviral Transduction of Bone Marrow-Derived DCs (BMDCs)
- Retrovirus Production: Clone cDNA into a retroviral vector (e.g., pMIG). Transfect Plat-E packaging cells using PEI. Collect virus-containing supernatant at 48 and 72h.
- BMDC Generation & Transduction: Flush bone marrow from femurs of C57BL/6 mice, lyse RBCs. Culture in RPMI + 10% FBS, 20 ng/mL GM-CSF. On day 3, add retroviral supernatant + 8 µg/mL polybrene, spinfect (2000g, 90 min, 32°C).
- Analysis: Continue culture in GM-CSF. After 48h, assess transduction efficiency via GFP reporter (if using pMIG) by flow cytometry. Sort GFP+ cells on day 6-7 for downstream phenotypic analysis.
Small Molecule Inhibition in Primary Immune Cells
Application: Acute inhibition of kinase signaling (e.g., BTK in B cells) for pathway dissection. Protocol: Dose-Response & Functional Assay
- Cell Preparation & Titration: Isolate primary human B cells via negative selection. Prepare a 10-point, 1:3 serial dilution of the inhibitor (e.g., Ibrutinib) in DMSO, with a final top concentration 10x the reported IC50.
- Pre-treatment & Stimulation: Plate cells at 1x10^5 cells/well. Add inhibitor or DMSO vehicle (≤0.1%) and pre-incubate for 1h. Stimulate cells with anti-IgM (10 µg/mL) to activate BCR signaling.
- Readout: After 15 min, lyse cells for phospho-protein analysis (p-BTK, p-PLCγ2) by Western blot or phospho-flow cytometry. After 48h, collect supernatant for IgG ELISA.
Visualizations
Perturbation Method Decision Tree
Mechanisms of Gene Perturbation
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Perturbation in Primary Immune Cells
| Reagent / Solution | Function & Application | Key Consideration for Immune Cells |
|---|---|---|
| Lentiviral dCas9-VPR/KRAB Systems | All-in-one vectors for stable, tunable gene activation/repression. | Low cytotoxicity and high titer are critical for sensitive primary cells (e.g., T cells). |
| Electroporation/Nucleofection Kits | Enable high-efficiency delivery of siRNA, plasmids, or RNP into hard-to-transfect cells. | Must be cell-type optimized (e.g., "Macrophage" or "T Cell" specific kits). |
| SMARTpool ON-TARGETplus siRNA | Pools of 4-5 siRNAs reduce off-target effects for more reliable knockdown. | Essential for primary cells where validation is resource-intensive. |
| Retroviral Vectors (e.g., pMIG, pMSCV) | Efficient gene transfer into dividing progenitor cells (e.g., bone marrow cultures). | Requires rapidly dividing target cells; ideal for ex vivo differentiated lineages. |
| Pathway-Specific Small Molecule Inhibitors/Activators | Acute, titratable, and often reversible modulation of specific protein targets. | Verify lack of off-target immune effects (e.g., on viability, baseline activation) via vehicle controls. |
| Recombinant Cytokines & Activation Beads | Maintain cell viability/phenotype and enable efficient transduction (activation). | Use human/mouse-specific, carrier-free cytokines at optimized concentrations. |
| Magnetic Cell Separation Beads | High-purity isolation of untouched immune cell subsets from primary tissue. | Negative selection avoids receptor cross-linking and unintended activation. |
| Low-Protein-Binding Plates/Tubes | Minimize adhesion and loss of low-abundance primary cells (e.g., monocytes). | Critical for accurate cell counts and functional assays post-manipulation. |
Comparative Analysis of Different CRISPRa/i Architectures for Immune Cell Applications
This Application Note is framed within a broader thesis on the use of CRISPR activation (CRISPRa) and interference (CRISPRi) for precise transcriptional modulation in primary immune cells, a critical capability for functional genomics and therapeutic development. The successful implementation of these technologies requires careful selection of the appropriate architecture tailored to the unique challenges of immune cell biology.
The performance, size, and delivery considerations of common CRISPRa/i systems are compared below for applications in primary T cells and macrophages.
Table 1: Comparative Performance of CRISPRa/i Architectures in Primary Immune Cells
| Architecture | Core Component | Approx. Size (kb) | Key Strength | Reported Gene Activation/Repression (Fold) | Primary Cell Suitability |
|---|---|---|---|---|---|
| dCas9-VPR | dCas9 + VP64-p65-Rta | ~9.5 | Strong, synergistic activation | Up to 2,000x (activation) | Good for T cells; can be stressful in some macrophages |
| dCas9-SunTag | dCas9 + scFv-GCN4 + VP64 | ~12.5 | Amplified signal, reduced payload per particle | Up to 1,500x (activation) | Excellent for T cells due to modularity |
| dCas9-SAM | dCas9-VP64 + MS2-P65-HSF1 | ~14.0 | Very strong, two-level recruitment | >2,000x (activation) | High efficiency in T cells; large size challenging for viral delivery |
| dCas9-KRAB | dCas9 + Krüppel-associated box | ~6.0 | Robust, stable repression | Up to 10-50x (repression) | Widely effective in immune cells; standard for CRISPRi |
| dCas9-DNMT3A/3L | dCas9 + DNA methyltransferase | ~11.0 | Epigenetic silencing | Up to 100x (repression via methylation) | Emerging use in macrophages for durable silencing |
Detailed Application Protocols
Protocol 1: Lentiviral Delivery of CRISPRa Systems into Primary Human T Cells
Objective: To achieve efficient transduction and transcriptional activation of an immunomodulatory target (e.g., IL2RA) in activated CD4+ T cells using the dCas9-VPR system.
Materials:
- Primary human CD4+ T cells, isolated and activated with CD3/CD28 beads.
- Lentiviral particles encoding dCas9-VPR and target-specific sgRNA (titer > 1x10^8 IU/mL).
- Polybrene (8 µg/mL final concentration).
- Complete T cell medium: RPMI-1640, 10% FBS, 100 U/mL IL-2.
- Flow cytometry antibodies for target validation (e.g., anti-CD25-APC).
Method:
- Day -1: Activate isolated CD4+ T cells with anti-CD3/CD28 Dynabeads (1 bead per cell) in complete medium + IL-2.
- Day 0: At 24h post-activation, spinoculate cells. Aliquot 1x10^6 cells per condition in a 24-well plate. Resuspend cell pellet in 1 mL of viral supernatant containing Polybrene. Centrifuge at 800 x g for 90 minutes at 32°C.
- Day 1: Carefully remove viral supernatant and resuspend cells in 2 mL of fresh complete medium + IL-2.
- Days 3-5: Assess transduction efficiency via dCas9-fluorescent protein reporter. Enrich if necessary.
- Day 6: Harvest cells and analyze target gene upregulation via flow cytometry (e.g., CD25 surface expression) and/or RT-qPCR. Compare to non-targeting sgRNA control.
Protocol 2: Electroporation of CRISPRi RNPs into Primary Human Macrophages
Objective: To achieve rapid, transient transcriptional repression of a cytokine gene (e.g., TNF) in monocyte-derived macrophages (MDMs) using dCas9-KRAB ribonucleoprotein (RNP) complexes.
Materials:
- Primary human monocytes differentiated to MDMs for 5-7 days with GM-CSF.
- Chemically synthesized sgRNA (with modifications for stability).
- Purified recombinant dCas9-KRAB protein.
- Electroporation buffer (P3 Primary Cell Solution, Lonza) and cuvettes.
- Nucleofector device (Lonza 4D-Nucleofector).
- LPS for stimulation post-editing.
Method:
- RNP Complex Formation: For one reaction, complex 6 µg (≈60 pmol) of dCas9-KRAB protein with a 1.2x molar excess of sgRNA (72 pmol). Incubate at room temperature for 15 minutes.
- Cell Preparation: Harvest MDMs using gentle cell dissociation reagent. Wash and count. Per reaction, pellet 5x10^5 to 1x10^6 cells.
- Electroporation: Resuspend cell pellet in 20 µL of P3 buffer. Mix with pre-formed RNP complex. Transfer to a nucleofection cuvette. Run the appropriate program (e.g., EH-100 for macrophages).
- Post-Electroporation: Immediately add 80 µL of pre-warmed culture medium. Transfer cells to a 24-well plate containing pre-warmed medium. Allow to recover for 24-48 hours.
- Stimulation & Analysis: Stimulate cells with 100 ng/mL LPS for 6 hours. Collect supernatant for TNF ELISA and cells for RNA analysis to quantify repression compared to non-targeting RNP control.
Visualizations
Title: CRISPRa Workflow for Primary T Cells
Title: Key CRISPRa/i Architectures and Function
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for CRISPRa/i in Immune Cells
| Reagent | Function & Application | Key Consideration |
|---|---|---|
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Production of high-titer, safe lentivirus for stable dCas9 delivery. | Essential for hard-to-transfect primary cells like resting T cells. |
| Recombinant dCas9-KRAB Protein | Enables rapid, transient CRISPRi via RNP electroporation. | Critical for minimizing off-target effects and toxicity in macrophages. |
| Chemically Modified sgRNA (e.g., 2'-O-methyl, phosphorothioate) | Increases nucleolytic stability and RNP formation efficiency in primary cells. | Markedly improves editing rates in immune cells. |
| IL-2 Cytokine | Maintains viability and proliferation of primary T cells post-transduction/activation. | Concentration must be optimized to balance survival and desired phenotype. |
| CD3/CD28 T Cell Activator Beads | Mimics antigen presentation to induce a replicative state permissive for lentiviral integration. | Critical pre-step for efficient T cell engineering. |
| Nucleofection Kit for Primary Cells (e.g., Lonza P3) | Specialized buffer/electroporation cuvettes for high-efficiency RNP delivery. | Cell type-specific programs are essential for viability. |
| Anti-sgRNA Negative Control | Non-targeting sgRNA matched in length/chemistry; critical for defining background signal. | Must be included in every experiment to validate on-target effects. |
Integrating CRISPRa/i with Single-Cell Multi-omics for Systems-Level Insights
This document provides a framework for using CRISPR activation (CRISPRa) and interference (CRISPRi) in tandem with single-cell multi-omics to dissect gene regulatory networks in primary immune cells. This integrated approach enables simultaneous perturbation and high-dimensional readout, moving beyond correlation to establish causality within complex cellular systems.
Key Applications:
- Functional Enhancer Mapping: Use CRISPRa/i to target candidate enhancers linked to disease-associated SNPs from GWAS and measure transcriptomic and epigenetic consequences in single cells.
- Regulatory Network Inference: Perturb key transcription factors or signaling nodes (e.g., STAT family proteins in cytokine responses) and use multi-omics readouts to reconstruct upstream and downstream regulatory cascades.
- Resistance Mechanism Elucidation: Perform CRISPRa/i screens on immune cells exposed to therapeutic agents (e.g., checkpoint inhibitors, cytokine therapies) to identify genes that confer sensitivity or resistance, coupled with deep phenotyping.
- Cell Fate Engineering: Activate or repress master regulator genes during differentiation (e.g., in CD4+ T cell subsets) and use multi-omics to trace divergent lineage trajectories and identify critical branching points.
Experimental Protocols
Protocol 1: Lentiviral Delivery of CRISPRa/i Systems into Primary Human T Cells
- Objective: Generate a stable pool of primary T cells expressing dCas9-effector proteins for perturbation studies.
- Materials: See "Research Reagent Solutions" table.
- Method:
- Virus Production: Co-transfect Lenti-X 293T cells with packaging plasmids (psPAX2, pMD2.G) and the transfer plasmid (e.g., lenti-dCas9-VPR for CRISPRa) using PEI transfection reagent. Harvest supernatant at 48h and 72h, concentrate via ultracentrifugation, and titer.
- T Cell Activation & Transduction: Isolate PBMCs from leukapheresis samples using Ficoll density gradient. Isolate CD3+ T cells via negative selection. Activate cells with CD3/CD28 Dynabeads (1:1 bead:cell ratio) in IL-2 (50 IU/mL) containing media.
- At 24h post-activation, transduce cells with lentivirus at an MOI of 5-10 in the presence of polybrene (8 µg/mL). Spinoculate at 800 x g for 90 min at 32°C.
- Selection & Expansion: At 72h post-transduction, begin selection with puromycin (1-2 µg/mL) for 7 days. Maintain cells in IL-2 media, expanding as needed for experiments. Validate dCas9 expression by flow cytometry or Western blot before use.
Protocol 2: Pooled CRISPRa/i Perturbation with Single-Cell Multi-omics Readout (CITE-seq)
- Objective: Screen a focused sgRNA library against immune checkpoints and transcription factors while capturing single-cell transcriptomes and surface protein abundances.
- Materials: See "Research Reagent Solutions" table.
- Method:
- Library Design & Cloning: Design a pooled sgRNA library (e.g., 3-5 sgRNAs/gene, 50-100 genes + non-targeting controls). Clone into a lentiviral sgRNA expression plasmid compatible with your dCas9-effector cell line.
- Large-Scale Transduction: Transduce your stable dCas9-expressing T cells (from Protocol 1) with the pooled sgRNA library at a low MOI (<0.3) to ensure single integrations. Use a representation of at least 500 cells per sgRNA.
- Perturbation & Stimulation: Culture transduced cells for 7-10 days to allow for transcriptional perturbation. Optionally, stimulate cells with PMA/ionomycin or anti-CD3/CD28 for the final 24h to reveal context-specific effects.
- Single-Cell Library Preparation: Harvest cells. Perform CITE-seq using the 10x Genomics Chromium Next GEM Single Cell 5' v3 kit. Stain cells with a TotalSeq-B antibody cocktail (e.g., CD3, CD4, CD8, CD45RA, CCR7, PD-1, CTLA-4) prior to loading. Generate libraries for gene expression, surface protein, and sgRNA amplicons per manufacturer's instructions.
- Sequencing & Analysis: Sequence libraries on an Illumina NovaSeq. Process data using Cell Ranger count with a custom reference including sgRNA sequences. Use Seurat for clustering and visualization. Assign perturbation identities via the sgRNA amplicon library (e.g., using Cellecta or MAGeCK methods). Perform differential expression (DE) and protein analysis per sgRNA cluster.
Table 1: Example Performance Metrics from Integrated CRISPRa/i + Single-Cell Multi-omics Experiments
| Metric | Typical Value/Outcome | Notes/Measurement Method |
|---|---|---|
| Lentiviral Titer on T Cells | 1-5 x 10^7 TU/mL | Measured by flow cytometry for a fluorescent reporter (e.g., GFP) 96h post-transduction. |
| Transduction Efficiency (Primary T cells) | 40-80% | Varies with donor and activation status. Critical for pool complexity. |
| Single-Cell Multiplexing Capacity | 5,000 - 10,000 cells/sample | Using standard 10x Genomics chips. |
| sgRNA Detection Rate | 20-50% of cells | Percentage of cells with confidently assigned sgRNAs from the amplicon library. |
| CRISPRa Fold Induction (Model Gene) | 5 - 50x | e.g., IL2RA (CD25) activation measured by surface protein (CITE-seq) vs. non-targeting control. |
| CRISPRi Knockdown Efficiency | 70-95% reduction | e.g., PDCD1 (PD-1) repression measured by transcript & protein vs. non-targeting control. |
| Differential Features per Perturbation | 50-500 genes | Number of significant (adj. p-value < 0.05) DE genes from a strong transcription factor perturbation. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Integrated CRISPRa/i - Multi-omics Workflows
| Item | Function | Example Product/Catalog # |
|---|---|---|
| dCas9-VPR Lentiviral Plasmid | Constitutive expression of CRISPRa effector (dCas9-VP64-p65-Rta). | Addgene #114189 (lenti dCas9-VPR) |
| dCas9-KRAB Lentiviral Plasmid | Constitutive expression of CRISPRi effector (dCas9-KRAB MeCP2). | Addgene #99378 (lenti dCas9-KRAB) |
| Pooled sgRNA Library Plasmid | Delivers guide RNA for specific gene targeting. Compatible with lentiviral production. | Custom designed, cloned into backbone like Addgene #84832 |
| CD3/CD28 T Cell Activator | Activates primary T cells, required for lentiviral transduction and expansion. | Gibco Dynabeads CD3/CD28, 11452D |
| Recombinant Human IL-2 | Supports survival and expansion of activated T cells. | PeproTech, 200-02 |
| Single-Cell 5' Kit w/ Feature Barcode | Enables simultaneous capture of transcriptome and surface protein (CITE-seq) data. | 10x Genomics, Chromium Next GEM Single Cell 5' v3, 1000269 |
| TotalSeq-B Antibody Cocktail | Antibodies conjugated to oligonucleotide barcodes for CITE-seq protein detection. | BioLegend, TotalSeq-B Human Universal Cocktail, 399906 |
| Chromium Controller | Microfluidic instrument for single-cell gel bead-in-emulsion (GEM) generation. | 10x Genomics, 1000204 |
| Cell Ranger Software | Primary analysis pipeline for demultiplexing, alignment, and feature counting. | 10x Genomics (open-source) |
| MAGeCK-FLUTE | Computational tool for analyzing CRISPR screen data, including single-cell modalities. | Open-source pipeline (Bioconductor) |
Diagram: Integrated Experimental Workflow
Workflow Title: CRISPRa/i Pooled Screen with CITE-seq Readout
Diagram: Signaling Pathway Analysis via Perturbation
Pathway Title: STAT3 Pathway Dissection via CRISPRa/i & Multi-omics
Conclusion
CRISPRa and CRISPRi have revolutionized the functional study of primary immune cells by enabling precise, scalable, and reversible transcriptional control. Moving beyond simple knockouts, these tools allow researchers to model disease states, dissect complex gene networks, and identify novel therapeutic targets with unprecedented precision. Successful implementation requires careful consideration of delivery methods, gRNA design, and rigorous validation to navigate the unique biology of primary cells. As delivery efficiency improves and next-generation effectors with enhanced specificity and reduced size emerge, the integration of CRISPRa/i screens with single-cell technologies will further accelerate discovery. The future points toward direct ex vivo and in vivo engineering of immune cells, paving the way for advanced cell therapies, personalized immunomodulation, and a deeper mechanistic understanding of immunity and immune-related diseases.