Prime Editing Explained: A Comprehensive Guide to the Next Generation of Precision Genome Engineering
This article provides a detailed exploration of prime editing, an advanced CRISPR-derived technology that enables precise genome modifications without double-strand breaks.
Prime Editing Explained: A Comprehensive Guide to the Next Generation of Precision Genome Engineering
Abstract
This article provides a detailed exploration of prime editing, an advanced CRISPR-derived technology that enables precise genome modifications without double-strand breaks. Tailored for researchers and drug development professionals, it covers the foundational mechanism of prime editors, their core components, and the step-by-step editing process. It further delves into methodological advancements from PE1 to PE7, therapeutic applications in diseases like Hurler syndrome and Tay-Sachs, and strategies to overcome challenges in efficiency and delivery. The content also includes a comparative analysis with base editing and CRISPR-Cas9, validating prime editing's superior precision and expanding clinical potential.
The Foundation of Prime Editing: From CRISPR-Cas9 to a Precision Gene Editor
The Limitation of First-Generation CRISPR and the Need for Precision
First-generation CRISPR-Cas9 technology revolutionized genetic engineering by providing unprecedented control over genome modification. However, this approach relies on creating double-strand breaks (DSBs) in DNA, which activates error-prone cellular repair mechanisms that frequently lead to unintended mutations [1]. These DSBs trigger predictable but problematic cellular responses: non-homologous end joining (NHEJ) often results in small insertions or deletions (indels) that disrupt gene function, while homology-directed repair (HDR) can produce precise edits but with relatively low efficiency in most cell types [2]. The consequences extend beyond simple inefficiency—DSB generation can activate the p53-mediated DNA damage response pathway, leading to cell cycle arrest, apoptosis, or cellular senescence [1]. Furthermore, chromosomal rearrangements, including translocations and large deletions, present significant safety concerns that substantially limit therapeutic applications [1]. These limitations highlighted an urgent need for more precise genetic modification technologies that could achieve predictable outcomes without inducing DNA breakage, spurring the development of precision editing tools like base editing and prime editing.
The Evolution of Precision Editing Tools
Base Editing: Foundations of Precision
Base editing emerged as the first major alternative to DSB-dependent editing, utilizing catalytically impaired Cas9 fused to nucleobase deaminase enzymes to enable direct chemical conversion of one DNA base to another without breaking the DNA backbone [1]. Cytosine base editors (CBEs) convert C•G to T•A base pairs, while adenine base editors (ABEs) convert A•T to G•C base pairs [3]. This approach significantly reduces indel formation compared to CRISPR-Cas9 nucleases and enables high-efficiency editing in both dividing and non-dividing cells [1]. However, base editing faces substantial constraints, including a narrow editing window (typically 4-5 nucleotides), strict PAM sequence requirements, and the potential for bystander edits where adjacent bases within the editing window are unintentionally modified [1]. Most significantly, base editors cannot achieve all 12 possible base-to-base conversions, nor can they perform targeted insertions or deletions, restricting their application scope [4]. These limitations prompted the development of a more versatile precision editing platform.
Prime Editing: A Search-and-Replace Mechanism
Prime editing represents a paradigm shift in precision genome editing by functioning as a "search-and-replace" system that directly writes new genetic information into a target DNA site without requiring DSBs or donor DNA templates [1] [4]. The core prime editing machinery consists of two primary components: (1) a prime editor protein formed by fusing a Cas9 nickase (H840A) to an engineered reverse transcriptase (RT), and (2) a specialized prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit [1] [4]. The editing process occurs through a coordinated multi-step mechanism beginning with target recognition and strand nicking, followed by reverse transcription of the edit-encoding RNA template, and culminating with DNA flap resolution and mismatch repair to permanently incorporate the genetic change [4]. This molecular architecture enables prime editing to perform all 12 possible base-to-base conversions, plus small insertions, deletions, and combinations thereof, dramatically expanding the range of addressable genetic variants compared to previous technologies [1] [4].
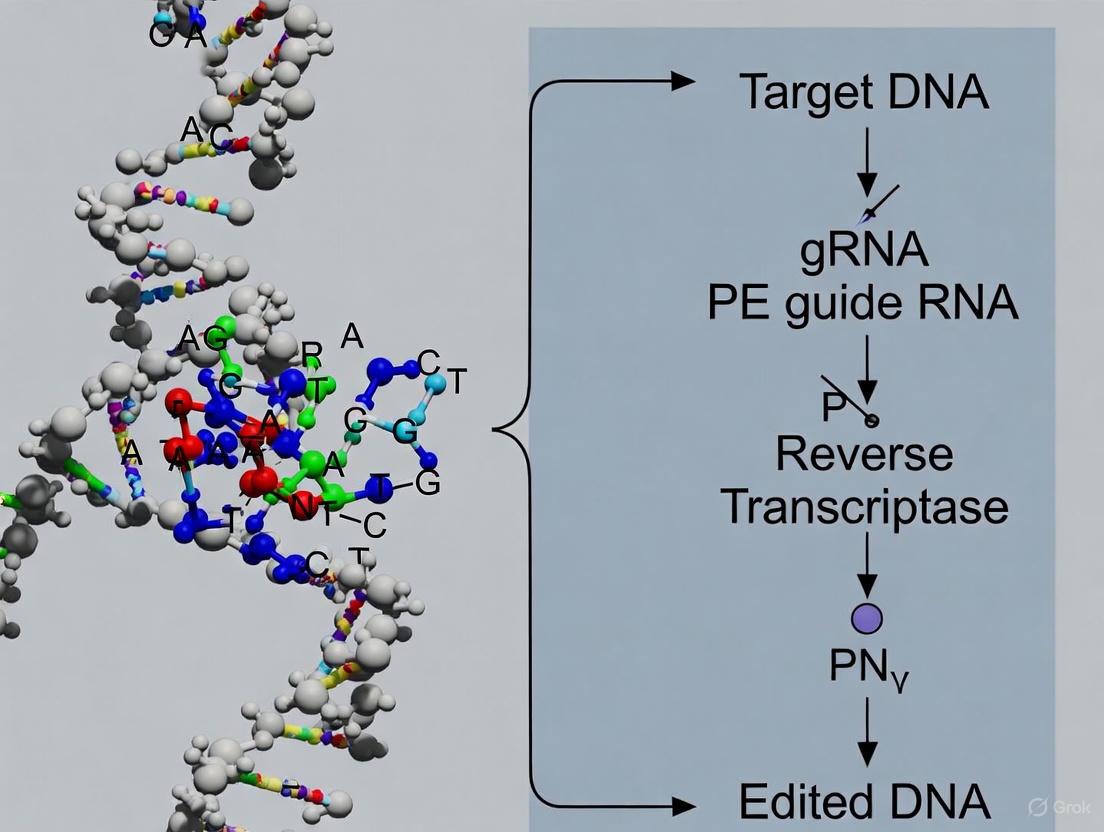
Prime Editing Mechanism: Molecular Workflow
The prime editing process operates through a precise biochemical pathway that can be visualized in the following workflow and described in detail thereafter:
Diagram 1: Prime editing molecular workflow. The process begins with complex assembly and proceeds through six key steps to achieve precise genome modification.
Component Architecture and Target Recognition
The prime editor fusion protein combines a Cas9 nickase (nCas9) with a reverse transcriptase (RT) domain, typically derived from Moloney murine leukemia virus (M-MLV) [5]. The nCas9 component contains a H840A mutation that inactivates the HNH nuclease domain, enabling only single-strand nicking rather than DSB formation [1] [4]. The pegRNA consists of four critical regions: (1) a spacer sequence that directs target recognition through standard Cas9:RNA DNA binding mechanics; (2) a scaffold region that facilitates Cas9 binding; (3) a primer binding site (PBS) that anneals to the nicked DNA strand; and (4) an RT template containing the desired edit [4] [3]. The initial binding event relies on canonical Cas9:gRNA DNA recognition, requiring a protospacer adjacent motif (PAM) sequence (typically 5'-NGG-3') adjacent to the target site [5].
DNA Nicking and Reverse Transcription
Upon binding to the target DNA, the nCas9 component introduces a single-strand nick at a specific position 3 nucleotides upstream of the PAM sequence on the non-target strand [5]. The newly liberated 3' DNA end then hybridizes with the complementary PBS sequence on the pegRNA, forming a primer-template complex that initiates reverse transcription [4]. The RT enzyme synthesizes DNA using the RT template region of the pegRNA, creating a complementary DNA flap containing the desired genetic edit [1]. This reverse transcription process continues until the entire edit sequence has been copied, resulting in a free 3' DNA flap that contains the newly written genetic information [4].
DNA Flap Resolution and Edit Incorporation
The newly synthesized edited DNA flap competes with the original 5' DNA flap for incorporation into the genome [4]. Cellular repair machinery recognizes and removes the unedited 5' flap while preferentially incorporating the edited 3' flap through a DNA ligation process [1]. This results in a heteroduplex DNA molecule with one strand containing the edit and the complementary strand retaining the original sequence [5]. The mismatch repair (MMR) system then recognizes this DNA mismatch and randomly resolves it using either strand as a template, resulting in permanent edit incorporation in approximately 50% of cases [1]. To bias this resolution toward the edited strand, the PE3 system introduces a second nicking guide RNA (ngRNA) that directs nCas9 to nick the non-edited strand, triggering repair that preferentially uses the edited strand as a template and increasing editing efficiency up to 4.2-fold [4].
Evolution of Prime Editing Systems
The rapid advancement of prime editing technology has produced multiple generations of editors with progressively improved capabilities, as summarized in the table below:
Table 1: Evolution of Prime Editing Systems
| Editor | Key Components | Editing Efficiency | Key Innovations | Applications |
|---|---|---|---|---|
| PE1 | nCas9 (H840A) + M-MLV RT | ~10-20% [1] | Proof-of-concept system | Initial demonstration of search-and-replace editing [1] |
| PE2 | nCas9 + engineered RT (5 mutations) | ~20-40% [1] | Optimized RT with enhanced processivity and stability [1] | Broad research applications; foundation for later systems [1] |
| PE3 | PE2 + additional sgRNA for non-edited strand nicking | ~30-50% [1] | Dual nicking strategy to bias MMR toward edited strand [1] [4] | High-efficiency editing in therapeutic contexts [1] |
| PEmax | Codon-optimized nCas9 (R221K/N394K) + optimized NLS | 50-95% (with MMR inhibition) [6] | Enhanced nuclear localization and expression [5] | High-efficiency editing in challenging cell types [5] |
| PE5 | PEmax + MLH1dn (MMR inhibition) | ~60-80% [1] | MMR suppression to prevent edit reversal [1] | Therapeutic applications requiring high editing rates [1] |
| PE6 | Compact RT variants (PE6a/b/c) or evolved M-MLV (PE6d) | ~70-90% [1] | Phage-assisted evolution for specialized editing tasks [1] [5] | Large insertions; challenging edits [1] [5] |
Later-generation editors have addressed specific limitations through creative molecular solutions. The PE4 and PE5 systems incorporate dominant-negative MLH1 (MLH1dn) to suppress MMR activity, significantly improving editing efficiency by preventing the removal of newly incorporated edits [1]. The PE6 platform represents a suite of editors featuring compact RTs (PE6a, PE6b, PE6c) for improved delivery or highly processive RTs (PE6d) for complex edits [1] [5]. Recent innovations have also produced Cas12a-based prime editors that recognize T-rich PAM sequences, expanding the targeting scope, and twinPE systems that use paired pegRNAs for larger edits [1] [7].
Experimental Framework for Prime Editing
Research Reagent Solutions
Table 2: Essential Reagents for Prime Editing Experiments
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Editor Proteins | PE2, PEmax, PE5, PE6 variants [1] [5] | Core editing machinery with varying efficiencies and specialties |
| Delivery Systems | Engineered virus-like particles (eVLP) [7], Lentiviral vectors [6], Lipid nanoparticles (LNPs) [3] | Cellular delivery of editing components |
| pegRNA Modifications | epegRNA (tevopreQ1 motif) [6], Circular pegRNA [1] | Enhanced pegRNA stability and editing efficiency |
| MMR Modulators | MLH1dn [1], Small molecule inhibitors | Suppress mismatch repair to improve editing outcomes |
| Cell Lines | MMR-deficient lines (e.g., MLH1-KO) [6], Stable editor-expressing lines [6] | Provide optimized cellular environment for editing |
| Analysis Tools | Next-generation sequencing, EditR, Primerize [8] | Quantify editing efficiency and specificity |
Protocol for High-Efficiency Prime Editing
The following experimental workflow illustrates the process for achieving high-efficiency prime editing based on established protocols:
Diagram 2: High-efficiency prime editing protocol. The seven-step workflow progresses from initial design to final validation of precise edits.
Step 1: Target Selection and pegRNA Design Identify target sites with appropriate PAM sequences (NGN for SpCas9-based editors) and design pegRNAs with systematic variation in PBS length (8-15 nt) and RT template length (10-30 nt) [6] [8]. Incorporate 3' structural motifs (e.g., tevopreQ1) to enhance pegRNA stability, creating engineered pegRNAs (epegRNAs) that significantly improve editing efficiency [6].
Step 2: Editor Selection Choose an appropriate prime editor based on the specific application: PE2 for basic editing, PEmax for general high-efficiency editing, PE5 when MMR antagonism is problematic, or PE6 variants for specialized tasks [1] [5]. For therapeutic applications requiring viral delivery, consider compact editors like PE6a or PE6b (1.2-1.5 kb) to accommodate packaging constraints [5].
Step 3: Delivery System Optimization Select delivery methods based on the target cell type and application. For research cell lines, lentiviral transduction provides efficient delivery and stable expression [6]. For therapeutic applications, engineered virus-like particles (eVLPs) offer transient expression with reduced immunogenicity [7]. Recent eVLP systems have achieved 65-fold improvements in delivery efficiency compared to earlier iterations [7].
Step 4: MMR Modulation Inhibit MMR activity to prevent edit rejection, either by using MMR-deficient cell lines (e.g., MLH1-KO) [6] or by co-expressing dominant-negative MMR proteins (e.g., MLH1dn in PE5) [1]. This critical step can improve editing efficiency up to 7-fold, particularly for small substitutions that MMR recognizes more efficiently [4].
Step 5: Stable Expression Establishment Generate cell lines with stable prime editor expression integrated into safe harbor loci (e.g., AAVS1) to enable continuous editing over multiple cell divisions [6]. Co-express fluorescent markers (e.g., EGFP) to track editor expression and facilitate cell sorting [6]. Allow extended editing periods (14-28 days) for edit accumulation, with efficiency monitoring at regular intervals [6].
Step 6: Editing Efficiency Quantification Harvest genomic DNA and amplify target loci using PCR, followed by next-generation sequencing to quantify precise editing rates and error profiles [6] [8]. Calculate precise editing efficiency as the percentage of sequencing reads containing only the intended edit without errors [6].
Step 7: Off-Target Analysis and Validation Perform whole-genome sequencing or targeted sequencing of predicted off-target sites to assess editing specificity [1] [4]. Compare editing outcomes with negative control cells to distinguish background mutations from true off-target events [6].
Current Limitations and Future Perspectives
Despite significant advances, prime editing faces several technical challenges that require further innovation. Delivery remains a primary constraint due to the large size of editor proteins and the complexity of pegRNAs, complicating packaging into delivery vectors with limited capacity, particularly adeno-associated viruses [4] [3]. Editing efficiency varies substantially across genomic loci and cell types, influenced by factors such as chromatin accessibility, DNA repair dynamics, and cellular state [8]. The MMR system continues to present a significant barrier, particularly for small edits that it efficiently recognizes and removes [6] [4]. Additionally, pegRNA design optimization remains partially empirical, requiring testing of multiple designs for each target to achieve optimal efficiency [8].
Future developments will likely focus on several key areas: (1) creating smaller editors through alternative Cas proteins and compact RTs to improve deliverability [1] [5]; (2) developing more sophisticated MMR evasion strategies through engineered editors or small molecule inhibitors [4]; (3) enhancing pegRNA stability and function through advanced chemical modifications and structural optimizations [6]; and (4) employing machine learning approaches to better predict editing outcomes based on sequence context and cellular environment [8]. As these technical hurdles are addressed, prime editing is poised to become an increasingly powerful tool for both basic research and therapeutic applications, potentially enabling correction of a broad spectrum of genetic mutations underlying human disease.
The transition from first-generation CRISPR to precision editing technologies represents a fundamental shift in our approach to genome engineering. By moving beyond double-strand breaks and their associated limitations, prime editing provides researchers with an unprecedentedly precise tool for writing genetic information. While challenges remain, the rapid evolution of this technology continues to expand its capabilities and applications, offering new pathways for understanding genetic function and developing transformative genetic medicines.
Prime editing represents a significant leap forward in the field of precision genome editing, enabling precise genetic modifications without inducing double-strand breaks (DSBs) or requiring donor DNA templates [9]. This technology has rapidly evolved into a versatile tool supporting a wide range of genetic modifications, including point mutations, insertions, and deletions. The core of this system is the prime editor protein complex, a sophisticated molecular machine that combines targeting and enzymatic functions to rewrite genetic information with exceptional accuracy. Understanding the architecture and function of this complex is fundamental for researchers and drug development professionals seeking to harness prime editing for therapeutic applications and basic research.
Architectural Composition of the Core Complex
Molecular Components and Their Functions
The prime editor protein complex is a multi-component system engineered to perform precise DNA editing through a coordinated mechanism. The core complex consists of a protein component and a specialized RNA guide that work in concert to identify target sequences and execute edits [9] [3].
Table 1: Core Components of the Prime Editing Complex
| Component | Type | Function | Key Features |
|---|---|---|---|
| Cas9 Nickase (H840A) | Protein | Binds and nicks target DNA | Creates single-strand break; Does not cause double-strand breaks [9] [3] |
| Engineered Reverse Transcriptase (RT) | Protein | Synthesizes DNA from RNA template | Fused to Cas9 nickase; Uses pegRNA template for DNA synthesis [9] [3] |
| Prime Editing Guide RNA (pegRNA) | RNA | Target recognition & edit template | Combines sgRNA spacer with RT template and primer binding site [9] [3] |
The fusion of a Cas9 nickase (H840A) with an engineered reverse transcriptase (RT) from Moloney Murine Leukemia Virus (MMLV) forms the protein backbone of the editor [9] [3]. This fusion creates a single polypeptide chain that can both locate a specific DNA sequence and catalyze the synthesis of new DNA at that site. The Cas9 nickase component is responsible for DNA binding and introduces a single-strand break in the non-target DNA strand, while the reverse transcriptase utilizes an RNA template to synthesize new DNA containing the desired edit.
Figure 1: Architecture of the prime editor complex, showing the fusion of Cas9 nickase and reverse transcriptase proteins guided by a multi-functional pegRNA.
The pegRNA: Blueprint and Guidance System
The prime editing guide RNA (pegRNA) is a critical component that distinguishes prime editing from other CRISPR-based systems. Unlike conventional single-guide RNAs (sgRNAs) used in standard CRISPR-Cas9 editing, the pegRNA serves dual functions: it directs the complex to the target DNA sequence and provides the template for the new genetic sequence to be written [3].
The pegRNA consists of four essential regions [3]:
- Spacer sequence: A ~20 nucleotide sequence that binds to the complementary target DNA locus, guiding the complex to the specific site for editing.
- Scaffold sequence: Enables binding to the Cas9 nickase protein and formation of the ribonucleoprotein complex.
- Reverse Transcription Template (RTT): Encodes the desired edit and provides flanking homology to facilitate the editing process, typically 25-40 nucleotides in length.
- Primer Binding Site (PBS): A 10-15 nucleotide sequence that anneals to the nicked DNA strand to initiate reverse transcription.
The extended length of pegRNAs (typically 120-145 nucleotides, but potentially up to 170-190 nucleotides) presents challenges for synthesis, delivery, and stability, which researchers must address through optimized design and delivery strategies [3].
Evolution of Prime Editing Systems
Development from PE1 to PE7
The prime editing system has undergone significant evolution since its initial development, with successive generations offering improved efficiency and versatility. The journey began with PE1, which established the foundational proof-of-concept by coupling nCas9 (H840A) with reverse transcriptase to mediate editing, though with relatively limited efficiency [9].
Table 2: Evolution of Prime Editor Systems
| System | Key Improvements | Typical Editing Efficiency | Applications |
|---|---|---|---|
| PE1 | Foundational system: nCas9 (H840A) + MMLV-RT | Low (Baseline) | Proof-of-concept edits [9] |
| PE2 | Engineered RT with enhanced thermostability and processivity | 1.7-7.6x PE1 (Human cells) | Broad range of precise edits [9] |
| PE3 | Additional nicking sgRNA to edit complementary strand | 1.4-8.2x PE2 (Human cells) | High-efficiency editing [9] |
| PE7 | Fusion with La protein + optimized pegRNA | 6.8-11.5x PE2 (Zebrafish) | In vivo therapeutic applications [10] |
PE2 incorporated an engineered reverse transcriptase with mutations that enhanced thermostability, processivity, and affinity for RNA-DNA hybrid substrates, resulting in significantly improved editing outcomes [9]. PE3 further augmented efficiency by incorporating an additional sgRNA that nicks the non-edited DNA strand, encouraging the cellular repair machinery to use the newly synthesized edited strand as a template, thereby increasing the likelihood of successful edit incorporation [9]. The most advanced systems, including PE7, represent substantial leaps forward through fusion with accessory proteins like La, which stabilizes the editing complex and enhances efficiency, particularly when combined with engineered "La-accessible" pegRNAs containing polyU motifs [10].
Engineering Advances in Protein Components
Recent engineering efforts have focused on optimizing the protein components to enhance editing precision and reduce unwanted byproducts. A significant concern with earlier prime editors was the potential for the nCas9 (H840A) to inadvertently generate double-strand breaks, leading to unwanted insertions or deletions (indels). This limitation has been addressed through additional mutations such as N863A, which significantly reduces the enzyme's ability to create DSBs while maintaining efficient target editing [9].
Further innovations include the development of split prime editors (sPE) that separate nCas9 and RT into independently functioning units [9]. This approach not only maintains high precision but also facilitates delivery via dual AAV vector systems, addressing a significant challenge in therapeutic applications. In vivo demonstrations have shown promising results, including successful editing of the β-catenin gene in mouse liver and correction of a mutation in a mouse model of type I tyrosinemia [9].
Quantitative Performance Data
Efficiency and Fidelity Metrics
The performance of prime editing systems is quantified through multiple parameters, including editing efficiency (percentage of target cells successfully edited), purity (percentage of desired edits among all editing outcomes), and error rate (frequency of unintended modifications).
Table 3: Performance Metrics of Advanced Prime Editing Systems
| System | Editing Efficiency | Error Rate | Experimental Context |
|---|---|---|---|
| PE2 | Baseline (varies by locus) | Not specifically quantified | Human cell lines [9] |
| PE7 with La-accessible pegRNA | Up to 15.99% (6.8-11.5x PE2) | Not specifically quantified | Zebrafish embryos (RNP delivery) [10] |
| vPE (Variant PE) | Not specified | 1.7% (1 in 101 edits) to 0.18% (1 in 543 edits) | Mouse and human cells [11] |
| PERT System | 20-70% of normal enzyme activity restored | No detected off-target edits | Human cell models of Batten, Tay-Sachs, and Niemann-Pick diseases [12] |
Recent advances have substantially improved both efficiency and fidelity. The development of vPE, which incorporates mutated Cas9 proteins that destabilize the old DNA strands during editing, has dramatically reduced error rates from approximately one error in seven edits to as low as one error in 543 edits for high-precision modes [11]. In therapeutic contexts, the PERT (prime editing-mediated readthrough of premature termination codons) system has demonstrated impressive efficacy in restoring protein function across multiple disease models with no detected off-target edits or significant transcriptomic changes [12] [13].
Experimental Workflows and Methodologies
Ribonucleoprotein (RNP) Complex Delivery
The delivery of prime editing components as pre-assembled ribonucleoprotein (RNP) complexes represents a highly efficient approach for introducing editors into cells, particularly for in vivo applications. The workflow involves several critical steps [10]:
Figure 2: Experimental workflow for prime editing using RNP complex delivery in zebrafish embryos, demonstrating a common approach for in vivo gene editing.
- RNP Complex Assembly: Purified PE7 protein is incubated with synthetic La-accessible pegRNA at specific concentrations (typically 750 ng/μL PE7 protein with 240 ng/μL pegRNA) to form functional RNP complexes [10].
- Microinjection: Approximately 2 nL of the RNP complex is delivered into the yolk cytoplasm of one-cell stage zebrafish embryos using microinjection techniques [10].
- Embryo Culture: Injected embryos are maintained at 28.5°C in a controlled environment to ensure proper development [10].
- Sample Collection: At 2 days post-fertilization (dpf), normally developed embryos are selected and collected for analysis [10].
- Genomic Analysis: DNA is extracted from pooled embryos (typically 6-8 per experimental group) using commercial kits, followed by target amplification with barcoded primers and next-generation sequencing to quantify editing efficiency [10].
This RNP delivery approach minimizes off-target effects and reduces potential immune responses compared to plasmid-based delivery methods, as the editing complexes are active for a shorter duration and degrade naturally within cells.
In Vivo Therapeutic Application
The PERT (Prime Editing-mediated Readthrough of Premature Termination Codons) system demonstrates a sophisticated therapeutic application of prime editing. This approach involves installing an engineered suppressor tRNA (sup-tRNA) to enable readthrough of premature stop codons, potentially treating multiple genetic diseases with a single editor [12] [13].
The experimental protocol involves [13]:
- tRNA Engineering: Screening tens of thousands of variants of all 418 human tRNAs to identify optimal sup-tRNA candidates through iterative optimization of leader sequences, tRNA sequences via saturation mutagenesis, and terminator sequences.
- Cell Model Validation: Testing optimized prime editors in human cell models of Batten disease (TPP1 p.L211X and p.L527X), Tay-Sachs disease (HEXA p.L273X and p.L274X), and Niemann-Pick disease type C1 (NPC1 p.Q421X and p.Y423X) using enzyme activity assays.
- In Vivo Testing: Delivering editors to mouse models of Hurler syndrome (IDUA p.W392X) via appropriate vectors and measuring protein restoration (approximately 6% IDUA enzyme activity) and pathological improvement.
This disease-agnostic approach demonstrates the potential for single editing compositions to treat multiple disorders caused by nonsense mutations, which account for approximately 24% of pathogenic alleles in the ClinVar database [13].
Research Reagent Solutions
Table 4: Essential Research Reagents for Prime Editing Experiments
| Reagent/Category | Function | Examples/Specifications |
|---|---|---|
| Prime Editor Plasmids | Express editor proteins in cells | PE2, PE3, PEmax, PE7 backbones [9] [10] |
| pegRNA Synthesis | Guide and template for editing | Chemically synthesized with 5'/3' modifications (methylated or phosphorothioate linkages) for stability [10] |
| La-accessible pegRNA | Enhanced stability and efficiency | pegRNA with 3' polyU motif for improved La protein binding [10] |
| Delivery Vectors | Introduce components into cells | Lipid nanoparticles (LNPs), AAV vectors (for split systems), electroporation systems [9] [3] |
| NGS Validation Kits | Quantify editing efficiency | Barcoded primers for amplicon sequencing; Illumina platforms [10] |
| Cell Culture Models | Test editing in human context | HEK293T, primary human fibroblasts, disease-specific cell models [9] [13] |
| Animal Models | In vivo functional testing | Zebrafish embryos, mouse disease models (e.g., Hurler syndrome) [13] [10] |
The research toolkit for prime editing has expanded significantly, with critical reagents spanning from editor expression constructs to specialized pegRNAs and delivery systems. Plasmid systems encoding various prime editor versions (PE2, PE3, PE7) are fundamental for initial testing and optimization [9] [10]. Specialized pegRNAs, particularly engineered versions with modified termini (evopreQ, mpknot, or polyU motifs), significantly enhance editing efficiency by protecting against degradation and improving protein binding [9] [10].
Delivery remains a crucial consideration, with lipid nanoparticles (LNPs) emerging as a promising vehicle for in vivo therapeutic applications, particularly for liver-targeted edits [9] [14]. For complex editors exceeding AAV packaging limits, split systems such as sPE (split prime editors) enable dual-vector delivery while maintaining functionality [9]. Validation reagents, including barcoded primers for high-throughput sequencing and specific antibody assays for functional validation, complete the essential toolkit for rigorous prime editing research.
Prime editing represents a transformative advancement in precision genome editing, enabling targeted installation of base substitutions, insertions, and deletions without requiring double-strand DNA breaks (DSBs) or donor DNA templates [9] [15]. At the heart of this versatile technology lies the prime editing guide RNA (pegRNA), a sophisticated molecular blueprint that directs both the targeting and the execution of precise genetic modifications [3]. Unlike conventional single guide RNAs (sgRNAs) used in CRISPR-Cas9 systems that merely specify the target location, pegRNAs additionally encode the desired edit and provide the necessary components for its implementation through reverse transcription [16] [3]. This multi-functional molecule has redefined the possibilities of genome engineering, offering researchers unprecedented control over genetic outcomes. The critical importance of pegRNA design and optimization cannot be overstated, as it directly determines the efficiency and success of prime editing experiments across diverse applications from therapeutic development to agricultural improvement [17] [18]. This technical guide deconstructs the pegRNA to provide researchers with a comprehensive framework for leveraging this powerful technology in their experimental systems.
Architectural Components of the pegRNA
The pegRNA comprises four essential sequence components that function in concert to enable precise genome editing. Each element serves a distinct purpose in the prime editing mechanism, and optimal design of each is crucial for achieving high editing efficiency.
Spacer Sequence: This 20-nucleotide region located at the 5' end of the pegRNA dictates target specificity by binding to the complementary DNA strand, guiding the prime editor complex to the intended genomic locus [3] [19]. The spacer sequence must be carefully selected to minimize off-target effects while maintaining strong on-target activity, with tools like the Doench 2016 score providing predictive efficiency metrics [19].
scaffold Structure: This section forms the stable secondary structure that binds the Cas9 nickase (nCas9) component of the prime editor, enabling its proper positioning and function at the target site [3]. The scaffold remains largely consistent with traditional sgRNA architectures but must accommodate the additional 3' extensions unique to pegRNAs.
Primer Binding Site (PBS): Typically 10-15 nucleotides in length, the PBS serves as an anchor point by binding to the 3' end of the nicked DNA strand, creating the primer-template complex that initiates reverse transcription [15] [20]. The PBS length and sequence significantly influence editing efficiency, with optimization required for different target contexts.
Reverse Transcription Template (RTT): This critical component encodes the desired genetic edit(s) and flanking homology region, serving as the template for the reverse transcriptase enzyme to synthesize the edited DNA strand [16] [3]. The RTT typically ranges from 25-40 nucleotides depending on the complexity of the intended edit, balancing the need for sufficient homology with synthetic constraints.
Table 1: Core Components of the pegRNA and Their Functions
| Component | Length (nt) | Primary Function | Design Considerations |
|---|---|---|---|
| Spacer | ~20 | Target recognition and binding | Minimize off-targets; maximize on-target efficiency |
| Scaffold | ~70-80 | Cas9 nickase binding | Maintain structural integrity for editor complex formation |
| PBS | 10-15 | Initiation of reverse transcription | Optimize length for binding strength and specificity |
| RTT | 25-40+ | Template for desired edit | Include edit with sufficient flanking homology |
The complete pegRNA molecule generally ranges from 120-145 nucleotides, significantly longer than standard sgRNAs, which presents both design and delivery challenges [3]. The extended length can complicate synthetic production, reduce cellular stability, and hinder delivery via viral vectors, necessitating specialized optimization strategies.
Molecular Mechanism of pegRNA-Directed Editing
The pegRNA orchestrates a sophisticated multi-step editing process through coordinated interactions with both the prime editor protein and the target DNA. Understanding this mechanism is essential for effective experimental design and troubleshooting.
Target Recognition and Complex Assembly
The process initiates with the formation of the prime editor-pegRNA complex, wherein the Cas9 nickase-reverse transcriptase fusion protein associates with the pegRNA scaffold region [3] [20]. The spacer sequence then directs this complex to the complementary target DNA site, with successful binding requiring both sequence complementarity and the presence of an appropriate protospacer adjacent motif (PAM) [19].
DNA Nicking and Primer Binding
Upon target binding, the Cas9 nickase component cleaves the PAM-containing DNA strand, generating a single-strand break with an exposed 3'-hydroxyl group [16] [20]. This nicked strand then serves as a primer for the subsequent reverse transcription step, with initiation dependent on hybridization between the primer binding site (PBS) of the pegRNA and the complementary sequence on the nicked DNA flap [15].
Reverse Transcription and Edit Incorporation
The reverse transcriptase enzyme utilizes the RNA-based RTT as a template to synthesize a new DNA strand extending from the primed 3' end, incorporating the desired edit specified in the RTT sequence [9] [3]. This results in a DNA heteroduplex intermediate containing both the original unedited strand and the newly synthesized edited strand, which exists as a 3' flap structure competing with the original 5' flap for integration [20].
Flap Resolution and Strand Correction
Cellular repair machinery processes the heteroduplex intermediate, preferentially removing the unedited 5' flap and ligating the edited 3' flap into the genomic DNA [15] [3]. For permanent incorporation of the edit in both DNA strands, additional systems like PE3 and PE3b introduce a second nick in the non-edited strand using a standard sgRNA, biasing cellular repair to use the edited strand as a template and resulting in a fully edited DNA duplex [16] [15].
Diagram 1: pegRNA Mechanism Flow - The sequential molecular steps of pegRNA-directed prime editing
Quantitative Design Parameters and Optimization Strategies
Systematic optimization of pegRNA components has yielded significant improvements in prime editing efficiency. The following parameters represent critical design considerations supported by empirical data.
Primer Binding Site (PBS) Optimization
The PBS length directly influences binding stability and reverse transcription initiation efficiency. Both excessively short and excessively long PBS sequences can impair editing efficiency, with optimal length depending on specific experimental context [20].
Reverse Transcription Template (RTT) Design
The RTT must balance two competing priorities: containing sufficient homology to facilitate proper flap equilibration and integration, while minimizing length to reduce synthetic complexity and potential degradation [15] [20]. Studies demonstrate that RTT length strongly influences editing efficiency, particularly in plant systems [20].
pegRNA Engineering for Enhanced Stability
A significant advancement in pegRNA technology came with the development of engineered pegRNAs (epegRNAs), which incorporate structured RNA motifs at the 3' end to protect against exonuclease degradation [9] [17]. These motifs include evopreQ1, mpknot, and Zika virus exoribonuclease-resistant RNA motifs (xr-pegRNA), which typically improve editing efficiency by 3-4-fold across diverse cell types [9].
Table 2: Experimentally Determined Optimization Parameters for pegRNA Components
| Parameter | Optimal Range | Impact on Efficiency | Validation |
|---|---|---|---|
| PBS length | 10-15 nt | Critical: <8nt insufficient binding; >16nt increased off-target effects | [15] [20] |
| RTT length | 25-40 nt | Strong effect: Shorter templates reduce efficiency; longer templates increase degradation | [15] [20] |
| RTT GC content | 30-70% | Moderate: Extreme values may impair functionality | [20] |
| epegRNA motifs | evopreQ1, mpknot | High: 3-4 fold improvement in mammalian cells | [9] [17] |
| Nicking sgRNA position | 40-100 nt from pegRNA nick | Variable: Dependent on edit type and cellular context | [15] |
Recent benchmarking studies utilizing stable expression of PEmax and epegRNAs in DNA mismatch repair-deficient cells have demonstrated remarkably high efficiencies, achieving up to 95% precise editing for certain targets [17]. These optimized conditions highlight the cumulative impact of synergistic improvements in pegRNA design, editor architecture, and cellular environment manipulation.
Experimental Protocol for pegRNA Design and Testing
This section provides a detailed methodology for designing, constructing, and validating pegRNAs for prime editing applications in mammalian cells, incorporating best practices from established protocols [15] [21].
pegRNA Design Workflow
Target Site Selection: Identify potential target sites adjacent to appropriate PAM sequences (5'-NGG-3' for standard Cas9) within 50 base pairs of the intended edit location [15] [19]. Verify target uniqueness using genome alignment tools to minimize off-target effects.
Spacer Design: Select a 20-nucleotide spacer sequence with high predicted on-target efficiency using established scoring algorithms (e.g., Doench 2016 score) [19]. Avoid spacers with potential off-target sites having fewer than 3 mismatches to the intended target.
PBS and RTT Determination: Design multiple pegRNAs (typically 3-5) with varying PBS lengths (10-15 nt) and RTT configurations for empirical testing [15] [21]. The RTT should encode the desired edit flanked by sufficient homology (typically 10-15 nt on each side) to support flap equilibration.
Nicking sgRNA Design (for PE3/PE5 systems): Design additional standard sgRNAs that target the non-edited strand, positioned 40-100 base pairs from the pegRNA-induced nick [15]. For PE3b systems, design nicking sgRNAs whose protospacer overlaps with the edit site to enhance specificity.
Diagram 2: pegRNA Design Workflow - Sequential process for designing and optimizing pegRNAs
pegRNA Cloning and Delivery
Molecular Cloning: Clone pegRNA expression cassettes into appropriate vectors using BsaI or BbsI restriction sites for golden gate assembly [21]. For epegRNAs, include structured RNA motifs (e.g., tevopreQ1) at the 3' terminus of the pegRNA scaffold.
Vector Selection: Utilize optimized prime editor systems such as PEmax (with codon-optimized RT and Cas9) for enhanced expression and nuclear localization [15] [17]. For challenging edits, consider PE4/PE5 systems that incorporate dominant negative MLH1 (MLH1dn) to suppress mismatch repair and improve efficiency [15] [17].
Cell Transfection: Deliver pegRNA and prime editor constructs to target cells using appropriate methods (electroporation for primary cells, lipid nanoparticles for difficult-to-transfect cells) [21] [3]. For stable expression systems, utilize lentiviral transduction with selection markers.
Efficiency Validation and Analysis
Next-Generation Sequencing: Assess editing efficiency 3-7 days post-transfection by PCR amplification of the target region followed by Illumina MiSeq sequencing [21]. Analyze sequence data for precise edits, indels, and other byproducts.
Clone Isolation and Characterization: For stable integration studies, isolate single-cell clones and expand for comprehensive genotyping via Sanger sequencing and functional validation [21].
Research Reagent Solutions for pegRNA Implementation
Successful implementation of prime editing requires access to specialized reagents and tools. The following table catalogues essential research reagents for pegRNA experimentation.
Table 3: Essential Research Reagents for pegRNA Experiments
| Reagent/Tool | Function | Example Sources |
|---|---|---|
| PEmax vector | Optimized prime editor with enhanced expression and nuclear localization | Addgene (#132775) [15] |
| pegRNA cloning vector | Backbone for pegRNA expression with U6 promoter | Addgene (#132777) [21] |
| epegRNA motifs | Structured RNA elements for pegRNA stabilization | [9] [17] |
| MLH1dn expression vector | Dominant negative MMR protein to enhance editing efficiency | Addgene (PE4/PE5 systems) [15] |
| p53DD expression vector | p53 dominant negative fragment to boost efficiency in hPSCs | Addgene (#41856) [21] |
| multicrispr R package | Computational tool for pegRNA design and off-target analysis | Bioconductor [19] |
The pegRNA represents both the targeting mechanism and the molecular blueprint for prime editing, integrating multiple functions into a single RNA molecule. Its sophisticated architecture—combining spacer, scaffold, PBS, and RTT components—enables precise "search-and-replace" genome editing without requiring double-strand breaks or donor DNA templates [16] [3]. While pegRNA technology has faced challenges related to variable efficiency and delivery, ongoing optimization strategies including epegRNA engineering, mismatch repair inhibition, and improved delivery systems have substantially enhanced its performance and reliability [9] [17] [18]. As prime editing continues to evolve toward therapeutic and agricultural applications, the pegRNA remains at the center of this revolutionary technology, offering researchers unprecedented precision in genome engineering. The design principles and experimental frameworks outlined in this guide provide a foundation for harnessing this powerful tool to address diverse genetic challenges.
The Fundamental 'Search-and-Replace' Mechanism
Prime editing represents a transformative advancement in genome engineering, offering unprecedented precision and versatility for genetic modifications. As a "search-and-replace" technology for the genome, it enables precise insertions, deletions, and all 12 possible base substitutions without requiring double-strand DNA breaks (DSBs) or external donor DNA templates [22] [23]. This groundbreaking approach significantly expands the therapeutic potential of genome editing while minimizing the risks of unintended mutations and genomic instability that can accompany conventional CRISPR-Cas9 systems [11].
The technology was developed to address critical limitations in existing genome-editing tools. While conventional CRISPR-Cas9 primarily generates insertions and deletions (indels) through DSB repair, and base editing is restricted to specific types of nucleotide substitutions, prime editing combines the programmability of CRISPR with the precision of reverse transcription to achieve a wider range of genetic modifications [22]. This versatility is particularly valuable for therapeutic applications, where precisely correcting pathogenic mutations requires high fidelity and minimal off-target effects. As noted by MIT researchers, "In principle, this technology could eventually be used to address many hundreds of genetic diseases by correcting small mutations directly in cells and tissues" [11].
Core Molecular Mechanism
The prime editing system consists of two primary components: a prime editor protein and a specialized prime editing guide RNA (pegRNA) [22] [23]. The editor protein is a fusion of a Cas9 nickase (nCas9) and a reverse transcriptase enzyme. The nCas9 component contains a H840A mutation that renders it capable of nicking only a single DNA strand, unlike wild-type Cas9 which creates double-strand breaks [23]. The reverse transcriptase domain, typically derived from Moloney murine leukemia virus (M-MLV RT), synthesizes DNA complementary to the RNA template provided by the pegRNA [22].
The pegRNA serves dual functions: it directs the nCas9 to a specific genomic locus and also templates the desired genetic modification [23]. Beyond the standard CRISPR guide RNA sequence that specifies the target site, the pegRNA contains two critical extensions at its 3′ end: a primer binding site (PBS) and a reverse transcription (RT) template encoding the desired edit [22] [23]. The editing process initiates when the nCas9 domain binds to the target DNA and nicks the strand containing the protospacer adjacent motif (PAM) [23]. The exposed 3′ end of the nicked DNA then hybridizes with the PBS region of the pegRNA, positioning it for reverse transcription [22]. The reverse transcriptase subsequently synthesizes a new DNA flap using the RT template as a guide, generating the programmed genetic modification [23]. Cellular repair mechanisms then resolve the resulting 3′ and 5′ flap structures, ultimately incorporating the newly synthesized DNA containing the desired edit into the genome [22].
Table 1: Core Components of the Prime Editing System
| Component | Description | Function |
|---|---|---|
| nCas9 (H840A) | CRISPR-Cas9 nickase mutant | Binds target DNA and nicks specific strand without creating double-strand break |
| Reverse Transcriptase | Typically M-MLV RT | Synthesizes DNA complementary to the RT template on the pegRNA |
| pegRNA | Engineered guide RNA with extensions | Specifies target site and templates desired genetic modification |
| PBS Sequence | Region of pegRNA | Anneals to nicked DNA strand to initiate reverse transcription |
| RT Template | Region of pegRNA encoding desired edit | Serves as template for DNA synthesis by reverse transcriptase |
Diagram 1: Prime editing molecular mechanism
Experimental Optimization and Validation
Efficiency Enhancement Strategies
Despite its precision, prime editing initially faced challenges with efficiency, prompting extensive optimization efforts. Recent approaches have focused on both the editing machinery and delivery methods. Systematic optimization has included structural and codon optimization of the nCas9-RT fusion enzyme, improvements to evade the mismatch repair pathway, and engineering more efficient pegRNAs [22]. Delivery method optimization has proven equally crucial, with the piggyBac transposon system emerging as an effective platform for stable genomic integration and sustained expression of prime editor components [22] [23]. This DNA-based transposition system facilitates gene transfer through a cut-and-paste mechanism, offering substantial cargo capacity (up to 20 kb) for multiplexed gene co-expression while circumventing immunogenicity concerns associated with viral delivery systems [22].
Combining these advancements has yielded remarkable improvements in editing efficiency. One comprehensive approach established single-cell clones with stable genomic integration of prime editors using piggyBac, utilized the CAG promoter for robust gene expression, and delivered engineered pegRNAs (epegRNAs) via lentivirus to ensure sustained expression for up to 14 days [22]. This integrated strategy achieved up to 80% editing efficiency across multiple cell lines and genomic loci, and demonstrated substantial efficiency (up to 50%) even in challenging human pluripotent stem cells in both primed and naïve states [22] [23].
Error Rate Reduction
Precision remains paramount in therapeutic genome editing. Recent research has addressed the error potential in prime editing, where the newly synthesized DNA flap must compete with the original DNA strand for incorporation into the genome [11]. If the original strand outcompetes the new one, the extra flap of new DNA may accidentally incorporate elsewhere, causing errors [11]. While early prime editors showed error rates ranging from approximately one error in seven edits to one in 121 edits for different editing modes, innovative protein engineering has dramatically improved fidelity [11].
MIT researchers developed modified Cas9 proteins with mutations that relax cutting constraints, making the original DNA strands less stable and more likely to be degraded [11]. This approach facilitates preferential incorporation of the new strands without introducing errors. By combining pairs of these mutations and incorporating them into a prime editing system with RNA binding proteins that stabilize the ends of the RNA template, researchers created a "vPE" editor that reduced error rates to just 1/60th of the original—ranging from one error in 101 edits to one in 543 edits across different editing modes [11].
Table 2: Prime Editing Efficiency and Error Rates Across Optimization Strategies
| Optimization Approach | Editing Efficiency | Error Rate | Application Context |
|---|---|---|---|
| PiggyBac + Lentiviral epegRNA | Up to 80% | Not specified | Multiple cell lines and genomic loci [22] |
| Stable Integration + CAG Promoter | Up to 50% | Not specified | Human pluripotent stem cells [22] |
| Original Prime Editor | Variable | 1:7 to 1:121 | Research settings [11] |
| vPE Editor | Maintained with improved precision | 1:101 to 1:543 | Mouse and human cells [11] |
Research Reagent Solutions
Successful implementation of prime editing requires carefully selected molecular tools and delivery systems. The following essential materials represent key components used in advanced prime editing experiments:
Prime Editor Plasmids: Engineered vectors such as pCMV-PE2 and pCMV-PEmax-P2A-hMLH1dn encode the fusion protein of nCas9 and reverse transcriptase [22] [23]. The PEmax variant represents a codon-optimized version with enhanced editing efficiency.
pegRNA Expression Systems: Specialized vectors (e.g., Lenti-TevopreQ1-Puro backbone) for cloning and expressing pegRNAs or epegRNAs (engineered pegRNAs) with structural modifications that improve stability and functionality [22] [23].
Delivery Vehicles:
- PiggyBac Transposon System: Comprises transposon plasmids with inverted terminal repeats (ITRs) and a transposase helper plasmid (e.g., pCAG-hyPBase) for precise genomic integration of prime editing components through cut-and-paste mechanism [22].
- Lentiviral Vectors: Used for delivering pegRNAs to ensure sustained expression in target cells [22].
Reporter Constructs: Validation tools such as mCherry-STOP-GFP reporters, where GFP expression occurs only after successful readthrough of premature termination codons, enabling quantitative assessment of editing efficiency [13].
Selection Markers: Fluorescent proteins (mCherry) or antibiotic resistance genes (Puromycin) enable enrichment of successfully transfected or transduced cells [22].
Diagram 2: Prime editing optimization framework
Therapeutic Applications and Case Studies
The "search-and-replace" capability of prime editing enables innovative approaches for treating genetic disorders. A notable therapeutic strategy is Prime Editing-mediated Readthrough of Premature Termination Codons (PERT), which addresses nonsense mutations that account for approximately 24% of pathogenic alleles in the ClinVar database [13] [24]. Rather than correcting individual mutations, PERT uses prime editing to permanently convert a dispensable endogenous tRNA into an optimized suppressor tRNA (sup-tRNA) that enables readthrough of premature stop codons [13].
This approach was validated in human cell models of Batten disease (TPP1 p.L211X and p.L527X), Tay-Sachs disease (HEXA p.L273X and p.L274X), and Niemann-Pick disease type C1 (NPC1 p.Q421X and p.Y423X), where treatment with the same prime editor installing an optimized sup-tRNA restored 20-70% of normal enzyme activity [13]. In vivo delivery of a single prime editor that converts an endogenous mouse tRNA into a sup-tRNA extensively rescued disease pathology in a Hurler syndrome model (IDUA p.W392X), with approximately 6% IDUA enzyme activity restoration sufficient to nearly eliminate disease signs [13]. This disease-agnostic strategy demonstrates how a single editing agent could potentially treat diverse genetic disorders caused by premature stop codons, benefiting patient populations with conditions including cystic fibrosis, Stargardt disease, phenylketonuria, and Duchenne muscular dystrophy [24].
Prime editing's fundamental "search-and-replace" mechanism represents a paradigm shift in genome engineering, combining precise targeting with versatile editing capabilities. Through continued optimization of both the editing machinery and delivery systems, researchers have achieved substantial improvements in efficiency and fidelity. The technology's ability to perform precise genetic modifications without double-strand breaks positions it as a powerful tool for both basic research and therapeutic development. As optimization efforts continue and delivery challenges are addressed, prime editing holds exceptional promise for treating a broad spectrum of genetic disorders through both mutation-specific correction and disease-agnostic approaches, potentially enabling a single therapeutic agent to benefit diverse patient populations.
Methodology and Therapeutic Applications: From Pipeline to Patient
Prime editing represents a transformative leap in genome editing technology, enabling precise modifications without introducing double-strand DNA breaks (DSBs) or requiring donor DNA templates [25] [1]. Developed in the lab of David Liu and first published in 2019, this "search-and-replace" editing system substantially expanded the scope of programmable genome editing beyond the capabilities of earlier technologies like CRISPR-Cas9 nucleases and base editors [26] [27]. The core innovation lies in fusing a catalytically impaired Cas9 nickase (nCas9) to a reverse transcriptase (RT) enzyme, programmed with a specialized prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit [25]. This review traces the systematic evolution of the prime editing platform from its initial proof-of-concept (PE1) to the highly optimized and specialized systems in use today (PE7), detailing the technical improvements that have enhanced its efficiency, precision, and applicability for therapeutic development and basic research.
The Fundamental Mechanism of Prime Editing
The prime editing mechanism distinguishes itself from previous CRISPR-based systems by directly writing new genetic information from a pegRNA template into a specific DNA locus. Figure 1 illustrates the multi-step process that underpins all prime editor versions.
Figure 1. The Core Prime Editing Mechanism. The process begins when the prime editor protein (nCas9-Reverse Transcriptase fusion) bound to a pegRNA locates and nicks the target DNA. The exposed 3' end hybridizes with the pegRNA's Primer Binding Site (PBS), initiating reverse transcription from the Reverse Transcriptase Template (RTT) that encodes the desired edit. Cellular machinery then resolves the resulting DNA flaps, and mismatch repair incorporates the edit permanently [25] [3] [27].
This mechanism enables the installation of all 12 possible base-to-base conversions, as well as small insertions and deletions, with high precision and minimal indel byproducts [26] [1]. Its versatility and safety profile make it particularly promising for correcting pathogenic mutations in a research and therapeutic context.
The Evolution of Prime Editing Systems
The journey from PE1 to PE7 is a story of continuous optimization, addressing key challenges such as low editing efficiency, pegRNA stability, and unproductive cellular repair responses. The major versions of prime editors and their defining characteristics are summarized in Table 1.
Table 1: Evolution and Key Characteristics of Prime Editing Systems
| System | Core Components & Modifications | Key Innovation | Typical Editing Efficiency* | Primary Applications & Advantages |
|---|---|---|---|---|
| PE1 [25] [1] | Cas9 H840A nickase + wild-type M-MLV RT | Proof-of-concept system | ~10–20% | Established the search-and-replace mechanism. |
| PE2 [25] [1] | Cas9 H840A nickase + engineered M-MLV RT (5 mutations) | Enhanced reverse transcriptase efficiency | ~20–40% | Higher efficiency across diverse sites; foundation for subsequent systems. |
| PE3 [25] [1] | PE2 + additional nicking sgRNA (ngRNA) | Dual-nicking strategy to bias repair | ~30–50% | Increased editing efficiency by guiding heteroduplex resolution. |
| PE3b [26] | PE2 + ngRNA designed to bind only the edited strand | Strand-selective nicking to reduce indels | Similar to PE3, with 13-fold fewer indels | Improved product purity by minimizing unwanted indel formation. |
| PE4 [26] [1] | PE2 + co-expressed dominant-negative MLH1 (dnMLH1) | Transient inhibition of mismatch repair (MMR) | ~50–70% | Boosted efficiency in MMR-proficient cell lines. |
| PE5 [1] | PE3 + co-expressed dnMLH1 | Combines dual-nicking with MMR inhibition | ~60–80% | Maximizes efficiency for challenging edits by addressing two bottlenecks. |
| PE6a-g [26] [1] | Variants with compact RTs (e.g., PE6a, PE6b) or evolved M-MLV RT/Cas9 domains | Specialized editors for different edit types and improved delivery | ~70–90% | AAV-compatible systems (compact variants); tailored solutions for specific edits. |
| PEmax [26] | Codon-optimized RT, additional NLS, mutations in Cas9 | Optimized protein expression and nuclear localization | Higher than PE2 | A improved "architecture" used as the base for further advanced systems. |
| PE7 [26] [1] | PEmax + fusion to La protein (PE7) | Enhanced pegRNA stability by blocking exonuclease degradation | ~80–95% | Improved performance, especially in challenging cell types. |
*Reported editing efficiencies are approximate and based on performance in HEK293T cells, as indicated in [1]. Efficiency is highly dependent on the specific target locus, cell type, and edit type.
Foundational Systems: PE1 to PE3
The first-generation systems established and refined the core prime editing workflow. PE1 demonstrated the feasibility of the technology but with modest efficiency, achieving editing at levels of 0.7–5.5% for point mutations and 4–17% for small indels [25] [27]. The key breakthrough came with PE2, which incorporated an engineered M-MLV reverse transcriptase with five mutations (D200N, L603W, T330P, T306K, W313F) that enhanced its thermostability, processivity, and DNA-RNA affinity, leading to a 2.3- to 5.1-fold average increase in efficiency over PE1 [25] [26] [27].
Building on PE2, the PE3 system introduced a second sgRNA to nick the non-edited strand. This additional nick encourages the cell's repair machinery to use the edited strand as a template, thereby increasing the likelihood of permanently incorporating the desired change and boosting efficiency by a further 2-3 fold [25] [1] [27]. A refined version, PE3b, uses an ngRNA designed to bind only after the edit has been made to the target strand, reducing the potential for undesired indels by 13-fold compared to PE3 [26].
Addressing Cellular Repair: PE4 and PE5
A significant barrier to efficient prime editing is the cellular mismatch repair (MMR) system, which can recognize the heteroduplex DNA (containing one edited and one original strand) and revert the edit to the original sequence [26]. The PE4 and PE5 systems were developed to overcome this barrier.
Both systems co-express a dominant-negative version of the MLH1 protein (MLH1dn) to transiently inhibit the MMR pathway [26] [1]. PE4 combines this MMR suppression with the PE2 editor, while PE5 combines it with the PE3 system. This strategic inhibition gives the edited DNA strand a greater chance to be permanently established, improving editing efficiency by 7.7-fold in PE4 versus PE2 and 2.0-fold in PE5 versus PE3 [26] [1].
Specialization and Delivery Optimization: PE6 and PEmax
Further optimization led to the development of the PE6 series and the PEmax architecture. The PEmax system features a codon-optimized reverse transcriptase for human cells, additional nuclear localization signals, and beneficial mutations in the Cas9 domain, collectively improving protein expression and activity [26].
The PE6 system, built upon PEmax, represents a move toward specialization. It includes a suite of editors (PE6a-g) developed through phage-assisted evolution [26]. Some variants (PE6a, PE6b, PE6c) use compact reverse transcriptase domains from bacterial retrons or retrotransposons, reducing the overall size of the editor to facilitate packaging into delivery vehicles like adeno-associated viruses (AAVs) [26] [1]. Other PE6 variants (PE6e-g) contain mutations in the Cas9 domain that can be combined with the evolved RTs for additive improvements in efficiency for specific types of edits [26].
Enhancing RNA Stability: PE7
A common issue with earlier systems was the degradation of the 3' extension of the pegRNA, which contains the critical PBS and RTT sequences. The PE7 system addresses this by fusing the PEmax editor to the La protein, a ubiquitous eukaryotic RNA-binding protein that stabilizes pegRNAs by protecting them from exonuclease degradation [26] [1]. This innovation enhances pegRNA stability and further boosts editing efficiency without requiring changes to the pegRNA design itself.
Experimental Protocols for Prime Editing
A typical prime editing experiment involves the careful design of editing components, their delivery into cells, and the analysis of outcomes. The following protocol outlines the key steps for implementing a PE3/PE5-type system in a mammalian cell line.
Component Design and Cloning
- pegRNA Design: The pegRNA must be designed to target the genomic locus and encode the desired edit. Tools like PrimeDesign (http://primedesign.pinellolab.org/) can automate this process, calculating parameters such as the PBS length (typically 10-15 nt) and RTT length (which includes the edit and necessary homology, often 25-40 nt) [28]. To enhance stability, engineered pegRNAs (epegRNAs) with 3' RNA pseudoknots can be used to prevent degradation [26].
- ngRNA Design: For PE3/PE5 systems, a nicking sgRNA (ngRNA) is designed to bind to the non-edited strand, approximately 50-100 bp away from the pegRNA nick site. The ngRNA should have a spacer sequence that matches the edited sequence to favor nicking only after successful editing (PE3b strategy) [26] [28].
- Plasmid Construction: The prime editor protein (e.g., PE2, PEmax) is typically encoded on one plasmid. The pegRNA and ngRNA are often cloned into separate expression plasmids under the control of U6 promoters [28]. The PE4/PE5 systems require an additional plasmid expressing the dominant-negative MLH1 [1].
Cell Transfection and Editing
- Cell Culture: HEK293T cells are a common model for initial testing. Cells are maintained in standard culture medium (e.g., DMEM with 10% FBS) and seeded into multi-well plates 18-24 hours before transfection to reach 70-80% confluency [28].
- Transfection: A common transfection mix for a 96-well format includes 30 ng of PE2/PE5max plasmid, 10 ng of pegRNA plasmid, and 3.3 ng of ngRNA plasmid, using a transfection reagent like TransIT-X2 [28]. For PE4/PE5, the MLH1dn plasmid must be included. The optimal ratio may require empirical adjustment for different cell lines or edits.
- Incubation: Cells are analyzed 72 hours post-transfection to allow sufficient time for editing and protein turnover [28].
Analysis of Editing Outcomes
- Genomic DNA Extraction: Cells are lysed, and genomic DNA is extracted using a commercial kit or a simple proteinase K-based lysis buffer [28].
- Amplicon Sequencing: The target locus is amplified by PCR, and the products are subjected to next-generation sequencing (NGS). This is the gold standard for quantifying prime editing efficiency and byproducts.
- Efficiency Calculation: NGS data is processed using computational pipelines (e.g., CRISPResso2) to calculate the percentage of reads containing the desired edit, as well as the rates of indels and other unintended modifications [28].
The Scientist's Toolkit: Essential Reagents for Prime Editing Research
Successful implementation of prime editing requires a suite of specialized reagents and tools. The following table details key resources for researchers.
Table 2: Essential Research Reagents and Tools for Prime Editing
| Item | Function & Importance | Examples & Notes |
|---|---|---|
| Prime Editor Plasmids | Expresses the fusion protein (nCas9-RT). The backbone for all editing activity. | - PE2/pJUL2440: A common PE2 construct with a P2A-eGFP reporter [28].- PEmax: An optimized backbone with improved expression [26]. |
| pegRNA Cloning Vectors | Allows for the expression of the complex pegRNA, which includes the spacer, scaffold, PBS, and RTT. | - pUC19-hU6-pegRNA-gg-acceptor: A common entry vector for pegRNA cloning (Addgene #132777) [28]. |
| ngRNA Cloning Vectors | For expressing the nicking sgRNA in PE3/PE5 systems. | - Standard U6-sgRNA expression vectors (e.g., Addgene #65777) [28]. |
| MMR Inhibition Plasmid | Expresses the dominant-negative MLH1dn protein to transiently suppress mismatch repair and boost efficiency. | - Required for the PE4 and PE5 systems [26] [1]. |
| Design Software | Critical for designing effective pegRNAs and ngRNAs by automating parameter optimization. | - PrimeDesign: A user-friendly web and command-line tool for designing single and pooled pegRNAs [28].- PrimeVar: A database of pre-designed pegRNAs for pathogenic ClinVar variants [28]. |
| Delivery Tools | Methods to introduce prime editing components into cells. | - Lipid Nanoparticles (LNPs): For non-viral delivery of RNP or mRNA [3].- Engineered Virus-Like Particles (eVLPs): For in vivo delivery with improved efficiency and safety [7].- Viral Vectors (AAV, Lentivirus): AAV is suited for in vivo delivery but has limited cargo capacity, favoring compact editors like PE6a-c [26] [1]. |
The evolution from PE1 to PE7 demonstrates a remarkable trajectory of scientific problem-solving, driven by a deep understanding of the biochemical and cellular barriers to precise genome editing. Each generation has systematically addressed a key limitation: reverse transcriptase efficiency (PE2), heteroduplex resolution (PE3), mismatch repair (PE4/PE5), editor size and specialization (PE6), and guide RNA stability (PE7). This continuous refinement has transformed prime editing from a novel proof-of-concept into a powerful and precise platform capable of correcting a vast majority of known pathogenic genetic variants [25] [1]. For researchers and drug development professionals, this expanding toolkit offers increasingly sophisticated and viable strategies to target the genetic root causes of diseases, paving the way for a new generation of therapeutic interventions.
This technical guide details the application of prime editing (PE) to correct disease-causing mutations in live animal models, situating these advances within the broader research on how prime editing works. It provides an in-depth analysis for researchers and drug development professionals, focusing on experimental outcomes, detailed methodologies, and key reagents.
Quantitative Analysis of In Vivo Prime Editing Outcomes
The following tables summarize key quantitative data from recent pioneering in vivo prime editing studies, providing a benchmark for editing efficiency and functional recovery.
Table 1: Prime Editing Efficiency in the TIGER Mouse Model Across Tissues [29]
| Tissue / Cell Type | Delivery Method | Editor | Editing Efficiency (%) | Readout Method |
|---|---|---|---|---|
| Retinal Pigment Epithelium | AAV | ABE / PE | Quantified via fluorescence restoration | In vivo & ex vivo confocal microscopy |
| Photoreceptors | AAV | ABE / PE | Quantified via fluorescence restoration | In vivo & ex vivo confocal microscopy |
| Müller Glia | AAV | ABE / PE | Quantified via fluorescence restoration | In vivo & ex vivo confocal microscopy |
| Trabecular Meshwork | AAV | ABE / PE | Quantified via fluorescence restoration | In vivo & ex vivo confocal microscopy |
| Hepatocytes | AAV (Intravenous) | PE | Quantified via fluorescence restoration | Confocal microscopy |
| Skeletal Muscle | AAV (Intravenous) | PE | Quantified via fluorescence restoration | Confocal microscopy |
| Brain Neurons | AAV (Intravenous) | PE | Quantified via fluorescence restoration | Confocal microscopy |
Table 2: Functional Rescue in Disease Models via the PERT Strategy [13] [12]
| Disease Model | Targeted Gene / Mutation | System | Functional Rescue | Pathology Rescue |
|---|---|---|---|---|
| Hurler Syndrome | IDUA p.W392X | Mouse Model | ~6% of normal IDUA enzyme activity | Nearly complete rescue of disease pathology |
| Batten Disease | TPP1 p.L211X, p.L527X | Human Cell Model | 20-70% of normal enzyme activity | N/D |
| Tay-Sachs Disease | HEXA p.L273X, p.L274X | Human Cell Model | 20-70% of normal enzyme activity | N/D |
| Niemann-Pick C1 | NPC1 p.Q421X, p.Y423X | Human Cell Model | 20-70% of normal enzyme activity | N/D |
| General Reporter | GFP nonsense mutation | Mouse Model | ~25% production of full-length GFP protein | N/A |
Experimental Protocols for Key In Vivo Studies
Protocol: Prime Editing in the TIGER Reporter Mouse
The TdTomato In Vivo Genome-Editing Reporter (TIGER) mouse model provides a versatile system for assessing prime editing delivery and efficiency across tissues with single-cell resolution [29].
- Animal Model: The TIGER mouse carries a constitutively expressed, mutated tdTomato gene (Q115X, Q357X) integrated into the Polr2a locus, abolishing fluorescence. Successful adenine base editing (ABE) or prime editing (PE) reverts the nonsense mutations and restores tdTomato fluorescence [29].
- Editor Delivery: Adeno-associated virus (AAV) vectors are packaged with the prime editing machinery and administered via local (e.g., intravitreal injection for ocular tissues) or systemic (intravenous injection) routes [29].
- Prime Editor Construct: The construct typically uses a PE2 system, which consists of a Cas9 nickase (H840A)-reverse transcriptase fusion protein and a prime editing guide RNA (pegRNA). For the TIGER model, pegRNAs were designed to correct Q115X or Q357X mutations using the PE2 approach without an additional nicking sgRNA [29].
- In Vivo Imaging and Analysis: Editing is quantified days to weeks post-injection using in vivo and ex vivo two-photon confocal microscopy to detect restored tdTomato fluorescence. Tissues can be processed for next-generation sequencing (NGS) to determine precise editing rates and indel frequencies [29].
Protocol: Disease-Agnostic Therapy with PERT
The Prime Editing-mediated Readthrough of Premature Termination Codons (PERT) strategy installs a suppressor tRNA (sup-tRNA) to read through nonsense mutations, offering a single therapeutic for multiple diseases [13] [12].
- sup-tRNA Engineering: A comprehensive screen of tens of thousands of variants of all 418 human tRNAs was conducted to identify sequences with high suppression potency. The optimized sup-tRNA was designed for the amber stop codon (TAG) [13].
- Genomic Installation via Prime Editing: A prime editor is programmed to permanently convert a single, dispensable endogenous tRNA gene (e.g., tRNA-Gln-CTG-6-1 or tRNA-Arg-CCG-2-1) into the optimized sup-tRNA. This avoids overexpression and preserves native regulation [13].
- In Vivo Validation:
- Reporter Mice: Mice are co-delivered with a reporter construct (e.g., GFP with a nonsense mutation) and the PERT editor. Successful editing is indicated by GFP expression [13].
- Disease Model (Hurler Syndrome): An IDUA p.W392X mouse model is treated systemically with the PERT editor. Functional rescue is assessed by measuring IDUA enzyme activity in tissues (e.g., brain, liver, spleen) and evaluating the amelioration of disease pathology [13] [12].
- Safety Profiling: Whole-genome sequencing (WGS) and transcriptomic/proteomic analyses are performed to assess off-target editing and global impacts on gene expression [13].
Visualizing Prime Editing Workflows and Strategies
The following diagrams illustrate the core mechanisms and experimental workflows for the in vivo success stories discussed.
Prime Editing Mechanism and PERT Strategy
Diagram 1: Core PE mechanism and the disease-agnostic PERT strategy. The standard prime editing process (solid path) leads to direct gene correction. The PERT strategy (dashed path) installs a sup-tRNA that enables readthrough of premature termination codons (PTCs) systemically [13] [12].
TIGER Mouse Model Workflow
Diagram 2: The TIGER mouse model workflow for assessing prime editing delivery. The model starts with a non-fluorescent mutant reporter. After AAV-delivered prime editing, successful correction is directly quantified by restored red fluorescence and NGS [29].
The Scientist's Toolkit: Key Research Reagents
This table catalogs essential materials and their functions, as used in the featured in vivo studies.
Table 3: Essential Reagents for In Vivo Prime Editing Research
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| TIGER Mouse Model | In vivo reporter for quantifying editing delivery and efficiency with single-cell resolution across tissues [29]. | Benchmarking AAV capsids or LNP formulations for organ-targeted delivery [29]. |
| PERT Prime Editor | A specific prime editing system designed to install an optimized suppressor tRNA at an endogenous genomic locus [13] [12]. | Developing disease-agnostic therapies for nonsense mutations in models of Hurler syndrome, Batten disease, etc. [13] [12]. |
| EnginepegRNAs (epegRNAs) | pegRNAs with 3' structural motifs (e.g., tevopreQ1) that enhance RNA stability and increase editing efficiency [17]. | Used in high-efficiency editing platforms in cell lines and animal models to achieve >90% precise editing [17]. |
| AAV Vectors | Viral delivery vehicle for in vivo transport of genome editing components. Can be serotyped for tissue tropism [29] [30]. | Delivering prime editors to the mouse eye, liver, muscle, and brain [29]. |
| MLH1-Deficient Cell Lines | Mismatch repair (MMR)-deficient cells (e.g., PEmaxKO) that prevent correction of prime edits, dramatically boosting efficiency [17] [6]. | Initial screening and optimization of pegRNAs prior to in vivo testing [17]. |
| Computational Design Tools (e.g., PRIDICT) | Machine learning models that predict pegRNA efficiency, guiding optimal pegRNA design for a given target sequence [31]. | Pre-screening pegRNA designs in silico to select the most promising candidates for experimental validation [31]. |
The development of personalized genetic medicines faces a fundamental scalability problem: creating a unique therapeutic agent for each of the thousands of known pathogenic mutations is economically and logistically challenging [13] [12]. Among the more than 200,000 disease-causing mutations documented in the ClinVar database, approximately 24% are nonsense mutations [13] [12] [32]. These mutations introduce a premature termination codon (PTC) within the coding sequence of an mRNA transcript, leading to truncated, non-functional proteins that underlie diverse genetic disorders [13] [33]. Until recently, therapeutic strategies required developing distinct genome-editing treatments for each specific nonsense mutation, significantly limiting the practical application of gene-editing technologies for rare diseases [12].
The Prime Editing-mediated Readthrough of Premature Termination Codons (PERT) strategy represents a paradigm shift from this mutation-specific approach. Developed by researchers at the Broad Institute, PERT leverages the versatility of prime editing to install a universal suppressor tRNA (sup-tRNA) that enables cells to read through PTCs regardless of their genomic context [13] [12]. This disease-agnostic platform has the potential to treat multiple unrelated genetic conditions with a single therapeutic agent, substantially expanding the population that could benefit from a single gene-editing drug [32]. By addressing a common molecular pathology across diverse diseases, PERT circumvents the need to invest multiple years and millions of dollars developing individual treatments for each genetic variant [12].
Technical Foundation: Prime Editing and Suppressor tRNAs
Prime Editing Architecture and Mechanism
Prime editing represents a significant advancement beyond earlier genome-editing technologies like CRISPR-Cas9 nucleases and base editors. While nucleases induce double-strand breaks (DSBs) that can lead to unpredictable repair outcomes, and base editors are limited to specific base transitions and suffer from bystander editing, prime editing offers a more precise and versatile approach [9]. The core prime editing system consists of a nCas9 (H840A) nickase fused to an engineered reverse transcriptase (RT), programmed by a specialized prime editing guide RNA (pegRNA) [9].
The editing mechanism begins when the prime editor complex binds to the target DNA sequence directed by the pegRNA. The nCas9 component nicks the non-target DNA strand, exposing a 3'-hydroxyl group that serves as a primer for reverse transcription. The RT then uses the template sequence encoded in the pegRNA to synthesize a new DNA strand containing the desired edit. Cellular repair mechanisms subsequently resolve this branched intermediate, incorporating the edit into the genome without requiring donor DNA templates or creating DSBs [9]. This "search-and-replace" capability enables all 12 possible base-to-base conversions, targeted insertions, and deletions with high precision and minimal byproducts [9].
Suppressor tRNA Biology and Engineering Challenges
In normal protein synthesis, termination codons (UAA, UAG, or UGA) in mRNA signal the release of the completed polypeptide chain. Nonsense mutations create premature versions of these stop signals, causing translational abortion before a functional protein is synthesized [13] [33]. Suppressor tRNAs represent a natural biological workaround for this problem—these specialized tRNAs recognize termination codons not as stop signals but as codons encoding specific amino acids, allowing translation to continue through the PTC to produce full-length proteins [13] [12].
However, therapeutic application of sup-tRNAs faced significant challenges. Existing approaches using viral delivery or lipid nanoparticles typically required repeated administration throughout the patient's lifetime and showed only modest potency, often necessitating potentially toxic overexpression [13] [33]. Additionally, the determinants of sup-tRNA potency were incompletely understood, making it difficult to engineer highly efficient variants [13]. The PERT strategy addresses these limitations through a one-time prime editing installation of an optimized sup-tRNA that functions at endogenous expression levels without disrupting global translation [13].
The PERT Methodology: Implementation and Optimization
High-Throughput tRNA Engineering and Screening
The development of PERT began with systematic engineering of sup-tRNAs with sufficient potency to mediate efficient nonsense suppression even when expressed from a single genomic copy. Researchers conducted iterative screening of tens of thousands of variants across all 418 high-confidence human tRNAs to identify optimal candidates [13] [12]. This comprehensive approach involved three sequential optimization phases:
First, the 40-bp leader sequence of tRNAs was systematically modified to enhance functionality. Next, saturation mutagenesis of the tRNA sequence itself was performed to identify mutations that improve suppression efficiency. Finally, the terminator sequence was optimized to ensure proper processing and stability [13]. Through this rigorous screening process, the team developed a highly active TAG-targeting sup-tRNA that maintained efficiency even at sub-endogenous expression levels from a single genomic locus [13].
Prime Editing Installation of Optimized sup-tRNA
The PERT strategy employs prime editing to permanently convert a dispensable endogenous human tRNA into the optimized sup-tRNA, avoiding the need for overexpression that can perturb global translation [13]. The human genome contains substantial redundancy in its tRNA genes, with 47 isodecoder tRNA families comprising 418 high-confidence genes [13]. This redundancy allows conversion of a single endogenous tRNA to a sup-tRNA with minimal cellular impact, as entire tRNA families have been deleted in humans without apparent consequences [13].
The installation process involves designing prime editing reagents to convert the anticodon loops of selected endogenous tRNAs into sup-tRNAs. For example, researchers successfully converted endogenous tRNA-Gln-CTG-6-1 and tRNA-Arg-CCG-2-1 into sup-tRNAs with an average conversion rate of 29% (ranging from 19% to 37%) in HEK293T cells [13]. This single genomic modification equips cells with a permanent source of the engineered sup-tRNA, creating a sustainable cellular environment capable of reading through PTCs regardless of which gene contains the nonsense mutation [13] [12].
Figure 1: PERT Workflow Overview - The three-phase implementation of the PERT strategy from tRNA engineering through functional protein restoration.
Experimental Models and Therapeutic Efficacy
In Vitro Validation in Human Disease Models
The therapeutic potential of PERT was rigorously evaluated across multiple human cell models of genetic diseases caused by nonsense mutations. Researchers tested the same prime editing system programmed to install the optimized sup-tRNA in models of Batten disease (TPP1 p.L211X and TPP1 p.L527X), Tay-Sachs disease (HEXA p.L273X and HEXA p.L274X), and Niemann-Pick disease type C1 (NPC1 p.Q421X and NPC1 p.Y423X) [13]. In all cases, treatment with the identical PERT editor resulted in significant restoration of enzymatic activity, demonstrating the disease-agnostic capability of this approach [13] [12].
To quantify the broad applicability of PERT, the team experimentally tested its effectiveness against all clinically relevant TAG PTCs in the ClinVar database. The results demonstrated successful PTC readthrough for the vast majority of sequences tested, confirming that a single sup-tRNA can potentially address numerous different nonsense mutations across diverse genetic contexts [13]. This finding underscores the remarkable versatility of the PERT platform and its capacity to serve as a universal solution for TAG-type nonsense mutations.
Table 1: In Vitro Efficacy of PERT in Human Disease Models
| Disease Model | Gene Mutation | Enzyme Activity Restoration | Citation |
|---|---|---|---|
| Batten disease | TPP1 p.L211X | 20-70% of normal | [13] [12] |
| Batten disease | TPP1 p.L527X | 20-70% of normal | [13] [12] |
| Tay-Sachs disease | HEXA p.L273X | 20-70% of normal | [13] [12] |
| Tay-Sachs disease | HEXA p.L274X | 20-70% of normal | [13] [12] |
| Niemann-Pick disease type C1 | NPC1 p.Q421X | 20-70% of normal | [13] [12] |
| Niemann-Pick disease type C1 | NPC1 p.Y423X | 20-70% of normal | [13] [12] |
In Vivo Therapeutic Efficacy in Animal Models
The PERT strategy was further validated in robust animal models, demonstrating both proof-of-concept and therapeutic efficacy. Initial testing in mice with a co-delivered reporter construct containing a nonsense mutation in GFP showed that a single prime editor converting an endogenous mouse tRNA into a sup-tRNA mediated approximately 25% production of full-length GFP [13]. This confirmed that the installed sup-tRNA could functionally read through PTCs in living animals.
More significantly, PERT was evaluated in a mouse model of Hurler syndrome, a severe lysosomal storage disease caused by the premature stop codon IDUA p.W392X [13] [12]. Treatment with the PERT system restored approximately 6% of normal IDUA enzyme activity in the brain, liver, and spleen—tissues normally severely affected by the disorder [13] [12]. Remarkably, this relatively modest restoration of enzymatic activity was sufficient to nearly eliminate all signs of disease pathology, demonstrating the therapeutic potential of this approach for treating human genetic disorders [13] [12].
Safety and Specificity Profile
A critical consideration for any genome-editing therapeutic approach is its safety and specificity. Comprehensive analysis of PERT-treated cells and animals revealed no detected off-target edits, changes in normal RNA or protein production, or toxicity to the cells [13] [12]. Several biological mechanisms contribute to this favorable safety profile and explain why the sup-tRNA preferentially reads through PTCs rather than natural termination codons (NTCs).
First, the distribution of stop codons for PTCs differs from that of NTCs, particularly enhancing the safety profile of sup-tRNAs targeting the amber stop codon (TAG) [13]. Second, NTCs are often followed by redundant and diverse in-frame stop codons that prevent significant protein extension even if the primary stop codon is read through [13]. Third, the recruitment of polypeptide chain release factors to the 3' untranslated region near NTCs can outcompete sup-tRNAs [13]. Additionally, if translation continues past the NTC, the resulting aberrant RNA and protein products are targeted for degradation through the non-stop decay pathway [13].
The PERT strategy further enhances safety by maintaining the engineered sup-tRNA at endogenous expression levels rather than overexpressing it, minimizing potential disruption to global translation [13]. Transcriptomic and proteomic analyses confirmed that PERT did not cause significant changes to overall gene expression or protein synthesis patterns [13].
Research Toolkit: Essential Reagents and Methodologies
Table 2: Key Research Reagents and Solutions for PERT Implementation
| Reagent/Solution | Function/Description | Application in PERT |
|---|---|---|
| Prime Editor (PE2/PE3) | Fusion of nCas9 (H840A) and engineered reverse transcriptase | Installs sup-tRNA at endogenous loci; PE2 for standard editing, PE3 with additional nicking gRNA for enhanced efficiency [9] |
| Engineered pegRNAs | Modified guide RNAs with 3' extensions containing RT template | Directs prime editor to target locus and encodes sup-tRNA sequence; often stabilized with evopreQ or mpknot motifs [9] |
| sup-tRNA Library | Tens of thousands of variants of human tRNAs | Screening resource for identifying optimal sup-tRNA sequences with high suppression efficiency [13] |
| mCherry-STOP-GFP Reporter | Dual fluorescence reporter system | Quantifies PTC readthrough efficiency; GFP expression occurs only after successful suppression [13] |
| HEK293T Cells | Human embryonic kidney cell line | Initial validation platform for sup-tRNA screening and prime editing efficiency [13] |
| Hurler Syndrome Mouse Model | IDUA p.W392X mutation | In vivo therapeutic efficacy assessment for lysosomal storage disease [13] [12] |
Figure 2: Molecular Mechanism of PERT - Comparison of normal translation, disease state with premature termination codon, and PERT intervention using engineered suppressor tRNA.
Discussion and Future Directions
The PERT strategy represents a significant conceptual advance in therapeutic genome editing by shifting the paradigm from mutation-specific correction to disease-agnostic intervention. By targeting a common molecular pathology shared across numerous genetic conditions, PERT addresses the critical bottleneck in developing treatments for rare diseases, where patient populations for individual mutations are often too small to justify the substantial development costs [12] [33]. This approach could potentially transform the economic landscape of genetic medicine, making therapies feasible for conditions that would otherwise be commercially non-viable [32].
Future development of PERT will likely focus on several key areas. First, expanding the sup-tRNA repertoire to address all three stop codons (TAG, TAA, and TGA) would further broaden the therapeutic scope beyond the current focus on amber mutations [13]. Second, optimizing delivery systems—particularly for challenging target tissues like the central nervous system—will be essential for clinical translation [13] [12]. The modular nature of prime editing components enables exploration of various delivery strategies, including viral vectors, lipid nanoparticles, and novel formulations [9] [34]. Third, thorough investigation of long-term safety and stability of prime-edited cells in diverse tissue types will be necessary before clinical application [13] [34].
The PERT strategy also inspires the development of other disease-agnostic therapeutic platforms that address common pathological mechanisms rather than individual mutations. As David Liu notes, "If you don't have to target one mutation at a time, the size of the patient groups that could be treated with a single drug becomes much, much larger. We hope the result will be many more patients that benefit, as well as greater incentives to develop gene-editing drugs for rare diseases" [12]. This approach could potentially be extended to other common mutation types, such as splice-site defects or promoter mutations, further expanding the reach of therapeutic genome editing.
The PERT strategy exemplifies the innovative application of prime editing technology to overcome fundamental challenges in genetic medicine. By permanently installing an optimized suppressor tRNA into a dispensable endogenous locus, PERT creates a cellular environment capable of reading through premature termination codons regardless of their genomic context. This disease-agnostic approach has demonstrated significant protein restoration and phenotypic rescue in multiple disease models, including Batten disease, Tay-Sachs disease, Niemann-Pick disease type C1, and Hurler syndrome, using a single therapeutic agent.
The technical foundation of PERT—combining sophisticated tRNA engineering with precise genome editing—showcases how creative molecular design can address practical limitations in therapeutic development. As research continues to optimize and expand this platform, PERT represents a promising path toward making gene-editing treatments accessible to larger patient populations affected by diverse genetic disorders caused by nonsense mutations. This strategy not only advances the technical capabilities of genome editing but also offers a more sustainable model for developing treatments for rare diseases that have historically been neglected due to economic constraints.
Current Clinical Pipeline and Approved Trials
Prime editing represents a significant advancement in precision genome editing, offering the ability to directly write new genetic information into a specified DNA site without inducing double-strand breaks (DSBs) or requiring donor DNA templates [35] [36]. This "search-and-replace" technology utilizes a catalytically impaired Cas9 endonuclease (nickase) fused to an engineered reverse transcriptase, programmed with a prime editing guide RNA (pegRNA) that specifies both the target site and encodes the desired edit [4] [36]. Since its initial description in 2019, prime editing systems have evolved through several generations (PE1 to PE7) with progressive improvements in editing efficiency, product purity, and targeting scope [1] [35].
The transition of prime editing from preclinical research to clinical application represents a milestone in genetic medicine. The first clinical trial of a prime editing-based therapeutic initiated in 2024, marking the debut of this versatile technology in human medicine [37]. This review comprehensively summarizes the current clinical pipeline and approved trials for prime editing therapies, providing detailed methodologies, visualizations, and research resources for the scientific community.
Current Clinical Trial Landscape
The clinical application of prime editing is currently focused on a pioneering trial, with several other potential therapies in advanced preclinical development. The table below summarizes the key clinical and near-clinical prime editing programs.
Table 1: Current Prime Editing Clinical Pipeline and Key Preclinical Candidates
| Condition/Therapeutic Area | Target Gene | Edit Type | Delivery Method | Development Stage | Key Results/Objectives |
|---|---|---|---|---|---|
| Chronic Granulomatous Disease (CGD) [38] | NCF1 | Correction of delGT mutation | Ex vivo HSC editing | Phase 1/2 Trial (NCT...)* | 66% DHR+ neutrophils by Day 30; rapid engraftment; well-tolerated |
| Hurler Syndrome (Preclinical) [12] [13] | IDUA | tRNA modification for nonsense suppression | In vivo (mouse model) | Preclinical | ~6% enzyme activity restoration; near-complete pathology rescue |
| Batten Disease (Preclinical) [13] | TPP1 | tRNA modification for nonsense suppression | In vitro (human cells) | Preclinical | 20-70% normal enzyme activity restored |
| Tay-Sachs Disease (Preclinical) [13] | HEXA | tRNA modification for nonsense suppression | In vitro (human cells) | Preclinical | 20-70% normal enzyme activity restored |
| Niemann-Pick Disease Type C1 (Preclinical) [13] | NPC1 | tRNA modification for nonsense suppression | In vitro (human cells) | Preclinical | 20-70% normal enzyme activity restored |
| Hereditary Transthyretin Amyloidosis (Preclinical) [14] | TTR | Protein reduction | LNP (systemic) | Preclinical (IND-enabling) | >90% protein reduction sustained |
Note: Specific NCT number was not provided in the search results. DHR+ refers to dihydrorhodamine-positive neutrophils, indicating restored NADPH oxidase activity.
Analysis of Current Clinical Progress
The prime editing clinical landscape demonstrates several key trends:
- Disease Focus: Initial applications target monogenic diseases with clear genetic etiology, particularly rare genetic disorders with high unmet need [38] [14].
- Delivery Approaches: Both ex vivo (e.g., hematopoietic stem cells) and in vivo approaches are being pursued, with each presenting distinct technical considerations [38] [35].
- Therapeutic Strategies: Approaches include direct mutation correction, nonsense mutation suppression via engineered tRNAs, and therapeutic protein reduction [13] [14].
Detailed Clinical Trial Analysis: PM359 for CGD
Trial Design and Protocol
The first-in-human prime editing trial is evaluating PM359 for p47phox chronic granulomatous disease (CGD) [38]. This Phase 1/2, multinational study employs the following methodology:
- Patient Population: Adult and pediatric participants with p47phox CGD caused by the prevalent delGT mutation in NCF1 [38].
- Study Design: Open-label trial assessing safety, biological activity, and preliminary efficacy of a single administration of PM359 [38].
- Intervention: Myeloablative conditioning with busulfan followed by intravenous infusion of autologous CD34+ hematopoietic stem cells edited ex vivo with prime editors designed to correct the delGT mutation [38].
- Endpoints: Primary safety endpoints include incidence of adverse events. Efficacy endpoints include restoration of NADPH oxidase function measured by dihydrorhodamine (DHR) assay, neutrophil and platelet engraftment kinetics, and incidence of serious infections [38].
Clinical Results and Outcomes
Interim results from the first patient demonstrate:
- Efficacy: Treatment led to restoration of NADPH oxidase activity in 58% of neutrophils by Day 15 and 66% by Day 30, significantly exceeding the 20% threshold anticipated for clinical benefit [38].
- Engraftment: Rapid engraftment occurred with neutrophil recovery by Day 14 and platelet engraftment by Day 19 - approximately twice as fast as median engraftment with approved gene editing technologies [38].
- Safety: No serious adverse events related to PM359 were reported. Adverse events were consistent with those expected from myeloablative conditioning with busulfan [38].
The following diagram illustrates the clinical development pathway and therapeutic mechanism for PM359:
Diagram Title: PM359 Clinical Pathway and Endpoints
Prime Editing Mechanism and Methodology
Molecular Mechanism of Prime Editing
Prime editing functions through a complex multi-step process that enables precise genome modification:
- Target Recognition: The prime editor complex (Cas9 nickase-reverse transcriptase fusion) binds to target DNA directed by the pegRNA spacer sequence [4] [3].
- DNA Nicking: The Cas9 nickase introduces a single-strand break in the non-target DNA strand [4] [36].
- Reverse Transcription: The primer binding site (PBS) of the pegRNA hybridizes to the nicked DNA strand, serving as a primer for reverse transcription using the RT template encoding the desired edit [4] [3].
- Flap Resolution: Cellular repair machinery resolves the resulting DNA flap structure, incorporating the edited strand containing the new genetic information [4] [36].
- Complementary Strand Editing (PE3/PE3b): An additional sgRNA directs nicking of the non-edited strand to bias repair to use the edited strand as a template [1] [4].
The following diagram illustrates this molecular process:
Diagram Title: Prime Editing Molecular Mechanism
Evolution of Prime Editing Systems
Prime editors have undergone significant optimization since their initial development:
Table 2: Evolution of Prime Editing Systems
| System | Components | Key Improvements | Editing Efficiency | Applications in Research |
|---|---|---|---|---|
| PE1 [1] | nCas9 (H840A) + M-MLV RT | Proof-of-concept | ~10-20% in HEK293T | Initial validation |
| PE2 [1] | nCas9 + engineered RT | Enhanced RT processivity/ stability | ~20-40% in HEK293T | Improved base conversions |
| PE3/PE3b [1] [4] | PE2 + additional sgRNA | Nicking of non-edited strand | ~30-50% in HEK293T | Higher efficiency edits |
| PE4 [1] | PE2 + MLH1dn | MMR inhibition | ~50-70% in HEK293T | Reduced indel formation |
| PE5 [1] | PE3 + MLH1dn | MMR inhibition + strand nicking | ~60-80% in HEK293T | High-efficiency editing |
| PE6 [1] | Compact RT variants, epegRNAs | Enhanced delivery, pegRNA stability | ~70-90% in HEK293T | In vivo applications |
| PEmax [35] | Optimized nCas9-RT | Nuclear localization, codon optimization | Variable by cell type | Therapeutic development |
Advanced Applications: Disease-Agnostic Approaches
PERT: Prime Editing-Mediated Readthrough of Nonsense Mutations
A groundbreaking disease-agnostic approach called PERT (prime editing-mediated readthrough of premature termination codons) addresses nonsense mutations that account for approximately 30% of rare genetic diseases [12] [13]. The methodology involves:
- Target Identification: Screening of 418 human tRNA genes to identify redundant tRNAs suitable for conversion [13].
- tRNA Engineering: Iterative optimization of tRNA sequences through saturation mutagenesis to create highly efficient suppressor tRNAs (sup-tRNAs) [12] [13].
- Genomic Integration: Using prime editing to permanently convert a single endogenous, dispensable tRNA gene into an optimized sup-tRNA [13].
- Functional Validation: Testing the sup-tRNA in disease models to assess protein rescue and functional recovery [12] [13].
This approach has demonstrated therapeutic potential across multiple disease models:
- Batten, Tay-Sachs, and Niemann-Pick Diseases: Restored 20-70% of normal enzyme activity in human cell models [13].
- Hurler Syndrome: Achieved approximately 6% enzyme activity restoration in a mouse model, sufficient to nearly eliminate disease pathology [12] [13].
The following diagram illustrates the PERT mechanism and experimental workflow:
Diagram Title: PERT Platform Workflow and Applications
Research Reagents and Methodological Toolkit
Table 3: Essential Research Reagents for Prime Editing Applications
| Reagent / Tool | Function | Examples / Specifications | Applications |
|---|---|---|---|
| Prime Editor Constructs | Core editing machinery | PE2, PE3, PEmax, PE6 variants [1] [35] | All prime editing applications |
| pegRNA Expression Systems | Target specification & edit encoding | Modified sgRNA scaffolds, epegRNAs [1] [4] | Determines editing outcome |
| Delivery Vehicles | Cellular delivery of editing components | LNPs, AAV vectors, electroporation [14] [35] | In vivo and ex vivo applications |
| MMR Modulators | Enhance editing efficiency | MLH1dn, MSH2dn [1] [4] | PE4, PE5 systems |
| Validation Assays | Edit confirmation & functional assessment | NGS, Sanger sequencing, functional assays [13] [38] | Efficacy and safety assessment |
| Cell Models | Experimental testing systems | HEK293T, iPSCs, primary cells [1] [13] | Protocol optimization |
| Animal Models | In vivo validation | Mouse models of genetic diseases [12] [13] | Therapeutic efficacy studies |
Experimental Protocol: Prime Editing in Hematopoietic Stem Cells
The clinical protocol for PM359 development exemplifies an optimized ex vivo prime editing workflow:
- HSC Collection and Isolation: Leukapheresis followed by CD34+ cell selection using clinical-grade magnetic separation [38].
- Stimulation Culture: Brief ex vivo culture in cytokine-supplemented media to enhance editing efficiency [38].
- Prime Editor Delivery: Electroporation of ribonucleoprotein (RNP) complexes containing prime editor protein and synthetic pegRNA [38].
- Quality Control Assessments:
- Editing efficiency quantification via NGS
- Cell viability and potency assays
- Sterility testing
- Transplantation: Myeloablative conditioning followed by infusion of edited cells [38].
- Engraftment Monitoring: Peripheral blood sampling for DHR assays and quantitative PCR to track edited cell populations [38].
The clinical pipeline for prime editing therapies, though still in early stages, demonstrates remarkable potential for addressing genetic diseases. The first clinical trial of PM359 for CGD has established proof-of-concept for therapeutic prime editing in humans, showing promising efficacy and an acceptable safety profile [38] [37]. Beyond this pioneering application, disease-agnostic approaches like PERT offer the potential for single therapeutic agents to address multiple genetic disorders caused by nonsense mutations [12] [13].
Future development will likely focus on overcoming remaining challenges, particularly in vivo delivery efficiency and immune compatibility [35] [3]. Additionally, expansion into more common genetic disorders and exploration of combinatorial approaches with other therapeutic modalities will further broaden the clinical impact of prime editing. As the field matures, prime editing is poised to become a foundational technology in the precision medicine arsenal, potentially capable of addressing thousands of genetic mutations across diverse therapeutic areas.
Overcoming Technical Hurdles: Strategies for Optimizing Prime Editing Efficiency
Addressing pegRNA Design and Stability Challenges
Prime editing is a versatile "search-and-replace" genome editing technology that enables precise genetic modifications without inducing double-strand DNA breaks. A prime editor consists of a Cas9 nickase (H840A) fused to an engineered reverse transcriptase, programmed by a specialized prime editing guide RNA (pegRNA). The pegRNA not only directs the editor to the target genomic site but also encodes the desired edit within its 3' extension, which comprises a primer binding site (PBS) and a reverse transcription template (RTT). Despite its theoretical promise to correct ~90% of known pathogenic mutations, the efficiency of prime editing remains a significant bottleneck for widespread research and therapeutic application. The central challenge lies in the pegRNA itself: its complex secondary structure, susceptibility to degradation, and suboptimal design parameters substantially limit editing efficiency and reliability. This technical guide examines the primary design and stability challenges associated with pegRNAs and outlines established and emerging strategies to overcome them, providing researchers with practical methodologies to enhance prime editing outcomes.
pegRNA Design Optimization Strategies
The design of the pegRNA is arguably the most critical factor determining prime editing success. Unlike standard CRISPR sgRNAs, pegRNAs contain additional structural components that introduce complex folding behavior and design constraints.
Machine Learning-Guided pegRNA Design
Traditional pegRNA design tools relied on rule-based approaches, but recent advances leverage machine learning to predict editing efficiency with much greater accuracy. These models analyze thousands of pegRNA sequences and their corresponding editing outcomes to identify features correlated with high performance.
Table 1: Machine Learning Tools for pegRNA Design Optimization
| Tool Name | Underlying Technology | Key Features | Reported Performance Improvement |
|---|---|---|---|
| OPED (Optimized Prime Editing Design) [39] | Nucleotide language model with transfer learning | Interpretable model; broad applicability across edit types and positions | 2.2-82.9× higher editing efficiency compared to low-scoring designs [39] |
| PRIDICT2.0 [40] | Ensemble of attention-based bidirectional recurrent neural networks | Predicts efficiency for replacements, insertions, deletions up to 40 bp; accommodates silent bystander edits | N/A |
| ePRIDICT [40] | Gradient-boosting algorithm | Accounts for local chromatin environment effects on editing rates | N/A |
The OPED framework exemplifies this approach, employing a sophisticated architecture that processes raw nucleotide sequences of target DNA and pegRNA pairs through transformer and attention networks to automatically learn comprehensive representations without relying on manually predefined features [39]. When validated across multiple datasets, pegRNAs with high OPED scores consistently showed significantly increased editing efficiencies—by 2.2 to 82.9-fold compared to low-scoring designs [39].
The Mismatched pegRNA (mpegRNA) Strategy
A key limitation of conventional pegRNAs is the high complementarity between their protospacer sequence and 3' extension, which promotes secondary structure formation that obstructs proper interaction with the target DNA and Cas9 protein [41]. Additionally, after successful editing, the original pegRNA may retain sufficient complementarity to bind the edited locus, leading to repeated nicking and increased indel formation [41].
The mpegRNA approach introduces strategic mismatches within the protospacer region (typically at positions N3-N11, with optimal performance often between N6-N10) to reduce complementarity and prevent persistent activity [41]. Experimental data demonstrates that this strategy enhances editing efficiency by up to 2.3 times while reducing indel levels by 76.5% without compromising performance [41]. When combined with epegRNA architectures, efficiency gains can reach 14-fold [41].
EXPERT System for Expanded Editing Range
Canonical prime editors can only modify genomic sequences downstream of the pegRNA nick site, significantly limiting their editing window. The EXPERT (extended prime editor system) addresses this limitation through two key modifications [42]:
- An extended pegRNA (ext-pegRNA) with elongated and modified 3' extension
- An additional sgRNA (ups-sgRNA) targeting the upstream region of the ext-pegRNA
This configuration creates two nicks on the same DNA strand ("cis nicks") and enables upstream binding, allowing EXPERT to perform editing on both sides of the ext-pegRNA nick [42]. The system demonstrates remarkable capability in replacing sequences up to 88 base pairs and inserting sequences up to 100 base pairs within the upstream region, with average editing efficiency improvements of 3.12-fold (up to 122.1 times higher) compared to PE2 for large fragment edits [42].
pegRNA Stability Enhancement Methods
Beyond design optimization, pegRNA stability represents a critical challenge, particularly for delivery as synthetic RNA or ribonucleoprotein (RNP) complexes.
Engineered pegRNA (epegRNA) Architectures
The original pegRNA design has been substantially improved through the addition of stabilizing RNA motifs to the 3' terminus, creating engineered pegRNAs (epegRNAs). These motifs protect against exonuclease degradation and reduce intrusive RNA folding.
Table 2: Strategies for Enhancing pegRNA Stability and Efficiency
| Strategy | Mechanism of Action | Key Improvement |
|---|---|---|
| epegRNA [1] | Addition of evopreQ1 or other RNA motifs to 3' end | Protects against exonuclease degradation; improves stability |
| L-epegRNA [43] | Splint ligation production with chemical modifications | Enables generation of long (170-190 nt), chemically modified epegRNAs |
| Csy4-mediated processing [1] | Co-expression of Csy4 ribonuclease and pegRNA with Csy4 recognition site | Separates pegRNA components to reduce misfolded structures |
| xrRNA motif [1] | Incorporation of exoribonuclease-resistant RNA motifs | Blocks processive degradation by cellular exonucleases |
| La protein fusion [1] | Fusion of La protein to prime editor complex | Binds 3' poly-U tail to protect from degradation and facilitate folding |
Chemical Modification and Synthesis Advances
A significant breakthrough in pegRNA stability comes from advances in production methods. Conventional in vitro transcription struggles to produce long, chemically modified pegRNAs necessary for therapeutic applications. Recently, researchers developed an efficient splint ligation approach that achieves approximately 90% production efficiency for chemically modified pegRNAs (125-145 nt) and epegRNAs (170-190 nt), referred to as L-pegRNA and L-epegRNA [43].
This method enables precise incorporation of chemical modifications such as 2'-O-methyl and phosphorothioate analogs, which dramatically enhance RNA stability and reduce immunogenicity [43]. When delivered as RNP complexes, L-epegRNA demonstrates editing efficiency improvements of up to more than tenfold across various cell lines and human primary cells compared to unmodified epegRNA produced by in vitro transcription [43]. With RNA delivery, improvements reach several hundredfold [43].
Experimental Protocols and Workflows
Protocol for High-Efficiency pegRNA Design Using Computational Tools
For researchers designing pegRNAs for specific edits, the following workflow integrates the most advanced computational tools:
Input Preparation: Compile the wild-type target DNA sequence (approximately 100 bp flanking the edit site) and precisely define the desired edit (substitution, insertion, or deletion up to 40 bp).
PRIDICT2.0 Analysis [40]:
- Access the web tool at www.pridict.it or install locally for batch processing.
- Input target sequence and edit specification.
- Generate multiple pegRNA designs with varying PBS and RTT lengths.
- Introduce silent bystander edits if needed to enhance efficiency.
- Obtain efficiency predictions for each design.
ePRIDICT Chromatin Context Evaluation [40]:
- Submit top-ranked pegRNA designs from PRIDICT2.0 to ePRIDICT.
- Analyze how local chromatin environment affects predicted editing rates.
- Select designs with optimal combined scores.
mpegRNA Optimization [41]:
- Introduce strategic mismatches (typically at positions N6-N10) in the protospacer region of top designs.
- Select mismatch bases (G, T, A, or C) based on empirical data showing optimal performance.
Stability Enhancement:
- Append evopreQ1 or other stabilizing motifs to create epegRNA constructs.
- For therapeutic applications, consider employing L-epegRNA with chemical modifications via splint ligation.
Protocol for L-epegRNA Production via Splint Ligations
For production of chemically modified L-epegRNAs, the splint ligation method provides superior results compared to in vitro transcription [43]:
RNA Oligonucleotide Preparation:
- Chemically synthesize two RNA fragments: the primary guide RNA fragment (containing spacer, sgRNA scaffold, and partial RTT) and the 3' extension fragment (containing remaining RTT, PBS, and stability motif).
- Incorporate 2'-O-methyl and phosphorothioate modifications at terminal positions during synthesis.
Splint DNA Template Design:
- Design a complementary DNA splint oligonucleotide that bridges the two RNA fragments with approximately 10-15 bp of complementarity to each fragment.
Ligation Reaction:
- Combine RNA fragments with splint DNA in 1:1.5:1.5 molar ratio.
- Heat mixture to 65°C for 2 minutes and slowly cool to room temperature for hybridization.
- Add T4 DNA ligase and reaction buffer, incubate at 37°C for 2-4 hours.
Purification and Quality Control:
- Treat reaction with DNase I to remove splint DNA.
- Purify full-length L-epegRNA using denaturing PAGE or HPLC.
- Verify integrity and concentration by analytical PAGE and spectrophotometry.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Advanced Prime Editing Research
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Prime Editor Systems | PE2, PEmax, PE3/PE3b, PE4max/PE5max [1] | Core editing machinery with optimized reverse transcriptase and nuclear localization |
| pegRNA Design Tools | OPED, PRIDICT2.0, ePRIDICT [39] [40] | Computational prediction of pegRNA efficiency and optimization |
| Stabilized pegRNA Formats | epegRNA (evopreQ1 motif), L-epegRNA (chemically modified) [43] | Enhanced RNA stability for improved editing outcomes |
| Delivery Systems | piggyBac transposon (stable integration), lentiviral vectors, RNP complexes [23] | Efficient intracellular delivery of editing components |
| MMR Inhibition | MLH1dn (dominant-negative MLH1) [1] | Suppression of mismatch repair to enhance prime editing efficiency |
| Specialized Cell Lines | MMR-deficient lines, reporter lines (GFP/mCherry-based) [13] | Optimization and quantification of editing efficiency |
The challenges of pegRNA design and stability represent significant but surmountable hurdles in prime editing research. Through integrated approaches combining computational design optimization (OPED, PRIDICT2.0), strategic engineering (mpegRNA, EXPERT), and advanced stabilization methods (L-epegRNA), researchers can now achieve editing efficiencies that robustly support both basic research and therapeutic development. The experimental protocols outlined provide a concrete framework for implementation, while the comprehensive reagent toolkit offers essential resources for successful experimentation. As these technologies continue to mature, the systematic addressing of pegRNA design and stability challenges will undoubtedly expand the scope and impact of prime editing in genetic research and therapeutic development.
Mitigating Mismatch Repair (MMR) Antagonism
Prime editing is a "search-and-replace" genome editing technology that enables precise modifications without introducing double-strand DNA breaks (DSBs). Despite its versatility, prime editing efficiency can be highly variable across different cell types and genomic loci. A significant cellular determinant influencing this efficiency is the DNA mismatch repair (MMR) pathway, which actively antagonizes prime editing outcomes [44] [45]. The MMR system, a fundamental DNA repair mechanism, recognizes and corrects mismatched nucleotides that arise during DNA replication. The heteroduplex DNA intermediate formed during prime editing—where one strand contains the newly synthesized edit and the complementary strand retains the original sequence—is perceived by the cell as a replication error to be corrected [44] [46]. This recognition often leads to the excision of the edited strand and restoration of the original sequence, thereby reducing editing efficiency and potentially promoting undesired indel byproducts [44]. Understanding and mitigating this MMR antagonism is therefore critical for enhancing the precision and efficacy of prime editing for both research and therapeutic applications.
Mechanistic Insights: How MMR Inhibits Prime Editing
The prime editing process culminates in a heteroduplex DNA structure containing a mismatch. The MMR machinery, specifically the MutSα (MSH2-MSH6) and MutLα (MLH1-PMS2) complexes, is recruited to this site [45]. The prevailing model suggests that the nick present on the edited strand serves as a signal for the MMR system, directing it to excise and re-synthesize the nicked strand. Consequently, the edit is removed, and the original genetic information is restored [44] [46]. This process directly counteracts the goal of prime editing. Research using CRISPRi screens has definitively identified that knockdown of key MMR genes, such as MLH1 and MSH2, results in a significant increase in prime editing efficiency—by 2 to 17-fold across various edit types and cell lines—confirming their inhibitory role [44] [45]. The following diagram illustrates the competitive dynamics between prime editing and MMR.
Researchers have developed multiple strategies to circumvent MMR antagonism. The table below summarizes the primary approaches, their mechanisms, and their quantitative impact on editing efficiency.
Table 1: Strategies for Mitigating MMR Antagonism in Prime Editing
| Strategy | Mechanism of Action | Key Reagents/Proteins | Reported Impact on Efficiency | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| PE4/PE5 Systems [44] [26] | Co-expression of a dominant-negative MLH1 (MLH1dn) protein to transiently inhibit the MutLα complex. | PE2 or PE3 editor, MLH1dn expression construct. | PE4: 7.7x ↑ vs. PE2; PE5: 2.0x ↑ vs. PE3; 3.4x ↑ edit/indel ratio [44]. | Significant efficiency boost; improved outcome purity; transient effect. | Potential safety concerns with prolonged MMR inhibition [46]. |
| Silent Mutations [44] | Inclusion of additional silent mutations in the RT template to create multiple mismatches, evading MMR recognition. | pegRNA designed with extra synonymous edits near the primary edit. | Efficiency enhancement observed, though specific fold-change varies by locus [44]. | Does not require manipulation of cellular machinery; highly precise. | Requires careful pegRNA design; efficacy may be sequence-dependent. |
| MMR Gene Knockdown [45] | Genetic disruption (e.g., CRISPRi/k.o.) of core MMR genes like MLH1 or MSH2. | sgRNAs targeting MMR genes. | 2x to 17x increase across edit types and cell lines [45]. | Potentially potent and stable effect; useful for mechanistic studies. | Not suitable for therapeutics due to genomic instability risk [46]. |
| Exonuclease Enhancement (Exo-PE) [47] | Recruitment of a 5'-3' exonuclease to degrade the original 5' flap, favoring incorporation of the edited 3' flap. | Engineered PE with aptamer-recruited exonuclease (e.g., T5 exonuclease). | ~7x ↑ for insertions ≥30 bp compared to PE2 [47]. | Particularly effective for larger insertions; avoids direct MMR inhibition. | Increased system complexity. |
The evolution of prime editors has led to a family of specialized systems. The following table provides a consolidated overview of the major prime editing systems, highlighting their components and progression.
Table 2: Evolution of Prime Editing Systems
| System | Core Components (vs. Previous) | Primary Mechanism/Improvement | Typical Editing Frequency (HEK293T) |
|---|---|---|---|
| PE1 [1] [46] | nCas9(H840A)-wildtype M-MLV RT, pegRNA | Foundational proof-of-concept. | ~10–20% [1] |
| PE2 [1] [26] | nCas9(H840A)-engineered M-MLV RT, pegRNA | Optimized reverse transcriptase (pentamutant). | ~20–40% [1] |
| PE3 [1] [26] | PE2 + additional nicking sgRNA | Nicks non-edited strand to bias repair towards the edit. | ~30–50% [1] |
| PE4 [44] [1] | PE2 + MLH1dn | Transient MMR inhibition via dominant-negative MLH1. | ~50–70% [1] |
| PE5 [44] [1] | PE3 + MLH1dn | Combines non-edited strand nicking with MMR inhibition. | ~60–80% [1] |
| PEmax [44] [26] | Codop-optimized RT, improved NLS, Cas9 mutations (R221K/N394K) | Enhanced editor expression, nuclear localization, and activity. | Varies; used as a backbone for PE4/5max [44]. |
| PE6(a-g) [48] [1] | Evolved/compact RTs (e.g., Ec48, Tf1), evolved Cas9 variants | Specialized editors for different edit types (small size/high processivity). | ~70–90% [1] |
Detailed Experimental Protocols for Key Strategies
Implementing the PE4/PE5 System with MLH1dn
The PE4 and PE5 systems represent a direct and effective method to mitigate MMR antagonism through the transient co-expression of a dominant-negative version of the MLH1 protein [44].
Methodology:
- Plasmid Constructs:
- Prime Editor: Utilize a plasmid expressing the PE2 protein (nCas9(H840A)-M-MLV RT fusion). For enhanced performance, the PEmax variant is recommended [44] [26].
- pegRNA: Clone your target-specific pegRNA into an appropriate expression vector. The pegRNA should encode the desired edit within its RT template.
- MLH1dn: Use a plasmid expressing the engineered dominant-negative MLH1 (e.g., MLH1 with specific point mutations that disrupt its function in the MutLα complex) [44].
- Nicking sgRNA (for PE5 only): For the PE5 system, a second plasmid expressing an sgRNA to nick the non-edited strand is required.
Cell Transfection:
- Co-transfect the target cells with the three plasmid types: PE2 (or PEmax), pegRNA, and MLH1dn. For PE5, include the nicking sgRNA plasmid.
- Adhere to standard transfection protocols optimized for your cell type. A typical DNA mass ratio to start optimization is 1:1:1 (PE2:pegRNA:MLH1dn).
Analysis:
- Harvest cells 48-72 hours post-transfection.
- Extract genomic DNA and amplify the target locus by PCR.
- Quantify editing efficiency using next-generation sequencing (NGS) or high-resolution melt curve analysis. Compare results to a PE2- or PE3-only control to calculate the fold-increase in efficiency [44].
Programming Silent Mutations via pegRNA Design
This strategy evades MMR by making the heteroduplex intermediate less recognizable to the repair machinery.
Methodology:
- Target Analysis:
- Identify the primary edit and the surrounding genomic sequence, particularly focusing on the ~100 bp flanking the edit.
pegRNA Design:
- Design the RT template to include one or more additional, silent mutations (synonymous codon changes) within ~10-30 bases of the primary edit. The goal is to create multiple mismatches in the heteroduplex.
- Selection of Silent Mutations:
- Prioritize mutations that do not create or disrupt known regulatory elements (e.g., splice sites, transcription factor binding sites).
- Use codon usage tables for the target organism to select a synonymous codon that maintains protein function but alters the DNA sequence.
- Ensure the primer binding site (PBS) of the pegRNA is complementary to the genomic sequence immediately downstream of the nick site and is not occluded by the newly introduced mutations.
Validation:
- Test the efficacy of pegRNAs with and without the additional silent mutations in a PE2 system (without MMR inhibition).
- Compare the editing efficiency of the primary edit between the two pegRNA designs using NGS. A successful design will show a significant increase in efficiency with the silent mutations [44].
The Scientist's Toolkit: Essential Research Reagents
Successful experimentation in mitigating MMR antagonism relies on a core set of reagents. The table below details these essential materials and their functions.
Table 3: Essential Reagents for MMR Antagonism Research
| Reagent Category | Specific Examples | Function in Experimentation |
|---|---|---|
| Prime Editor Plasmids | PE2, PEmax, PE6 variants (Addgene) [26]. | Engineered fusion protein that nicks DNA and reverse transcribes the edit from the pegRNA. |
| pegRNA Expression Vectors | plasmids with U6 promoter for pegRNA expression [44]. | Delivers the pegRNA, which specifies the target locus and encodes the desired edit(s). |
| MMR Inhibitor Constructs | Plasmid expressing dominant-negative MLH1 (MLH1dn) [44] [46]. | Transiently inhibits the MMR pathway to prevent rejection of the prime edit. |
| Nicking sgRNAs | sgRNA expression plasmid for PE3/PE5 systems [44] [26]. | Introduces a nick in the non-edited strand to bias cellular repair towards the edited strand. |
| Control Cell Lines | MMR-proficient (e.g., HEK293T, HeLa) vs. MMR-deficient (e.g., HCT116, MLH1-/- lines) [44] [45]. | Essential controls for benchmarking the effect of MMR inhibition strategies. |
| Editing Detection Tools | NGS platforms, T7 Endonuclease I assay, restriction fragment length polymorphism (RFLP). | For accurate quantification of editing efficiency and indel byproducts. |
Safety and Practical Considerations
While inhibiting MMR significantly enhances prime editing efficiency, it must be approached with caution. The MMR pathway is a critical guardian of genomic integrity. Its prolonged inactivation is associated with a hypermutator phenotype, characterized by microsatellite instability (MSI) and a significantly elevated risk of cancer development [46]. Therefore, transient inhibition—achieved by the short-term expression of MLH1dn in the PE4/PE5 systems—is considered a safer approach for research and potential ex vivo therapeutic applications compared to permanent genetic knockout of MMR genes [44] [46]. The strategy of using silent mutations presents no such safety concerns, as it works with the cellular machinery rather than against it. For therapeutic development, the balance between achieving high editing efficiency and minimizing oncogenic risk is paramount and requires careful evaluation.
Navigating Delivery Challenges for Large Editor Complexes
Prime editing represents a transformative advance in precision genome editing, enabling targeted insertions, deletions, and all 12 possible base-to-base conversions without double-strand DNA breaks [9] [26]. This technology combines a Cas9 nickase (H840A) fused to a reverse transcriptase enzyme, programmed by a specialized prime editing guide RNA (pegRNA) that specifies both the target site and the desired edit [9] [3]. While this sophisticated molecular machinery offers unprecedented editing precision, its substantial size creates significant delivery challenges that limit therapeutic application [9]. The prime editor protein and extended pegRNA collectively exceed the packaging capacity of standard adeno-associated virus (AAV) vectors, which have a ~4.7 kb limit [9] [26]. This review examines current strategies to overcome these delivery constraints while maintaining editing efficiency.
Structural Constraints and Size Limitations
The fundamental delivery challenge stems from prime editing's multi-component architecture. The system includes a fusion protein of nCas9 (H840A) and an engineered reverse transcriptase, along with an extended pegRNA typically 120-145 nucleotides long [3]. The pegRNA contains not only the standard CRISPR targeting spacer and scaffold, but also a primer binding site (PBS) and reverse transcription template (RTT) encoding the desired edit [9] [49].
This complex exceeds the packaging capacity of single AAV vectors, necessitating creative solutions for in vivo delivery [9]. Additionally, the large size and complexity of pegRNAs presents challenges for chemical synthesis, cellular stability, and delivery efficiency [3]. The 3' extension of conventional pegRNAs is particularly prone to degradation, reducing editing efficiency [9]. These molecular constraints have driven innovation in both editor engineering and delivery methodology.
Engineering Strategies to Reduce Editor Size
Split Prime Editing Systems
The most direct approach to overcoming size limitations involves splitting the prime editor into separate components that reassemble inside target cells. The split prime editor (sPE) system separates the nCas9 and reverse transcriptase domains, allowing them to function independently while maintaining editing precision [9]. This separation enables delivery via dual AAV vectors while avoiding increased indel mutations [9]. In proof-of-concept studies, sPE successfully edited the β-catenin gene in mouse liver and corrected a mutation in a mouse model of type I tyrosinemia using a dual AAV vector system [9].
Compact Editor Variants
Recent protein engineering efforts have yielded smaller prime editing systems with improved packaging potential. The PE6 series includes editors with reverse transcriptase domains derived from bacterial retrons (PE6a) and retrotransposons (PE6b), resulting in smaller fusion proteins [26]. PE6c and PE6d variants further evolved these compact enzymes to maintain efficiency for longer, more complex edits while remaining compatible with AAV delivery [26]. The development of these specialized editors represents a significant advance toward therapeutic application.
pegRNA Optimization and Stabilization
pegRNA instability presents another critical delivery challenge. Multiple approaches have emerged to address this limitation:
Engineered pegRNAs (epegRNAs) incorporate structured RNA motifs (evopreQ, mpknot, or G-quadruplex) at the 3' end to protect against degradation [9] [17]. These modifications improve editing efficiency 3-4-fold across multiple human cell lines without increasing off-target effects [9].
PE7 system utilizes fusion with the endogenous La protein, a small RNA-binding exonuclease protection factor that stabilizes pegRNA 3' ends [26]. This approach leverages natural RNA protection mechanisms to enhance prime editing efficiency.
Table 1: Prime Editor Systems and Their Delivery Characteristics
| Editor System | Key Components | Size Considerations | Delivery Compatibility |
|---|---|---|---|
| PE2 | nCas9-H840A + engineered RT | Large fusion protein | Plasmid, mRNA + pegRNA |
| PE3/PE3b | PE2 + nicking sgRNA | Additional sgRNA increases payload | Multiple vectors needed |
| PEmax | Codon-optimized PE2 + NLS | Improved nuclear localization | Enhanced non-viral delivery |
| sPE | Split nCas9 and RT | Enables dual-vector delivery | Dual AAV vectors |
| PE6a-d | Compact RT domains | Reduced size variants | Single AAV for some variants |
Delivery Methodologies for Prime Editing Systems
Viral Vector Strategies
Dual AAV Systems: Split prime editors can be packaged into separate AAV vectors, typically employing dual promoters or intein-mediated trans-splicing approaches [9]. This strategy successfully demonstrated therapeutic efficacy in mouse models, though editing efficiencies vary based on tissue type and transduction efficiency.
Single AAV Systems: For smaller prime editors like certain PE6 variants, extensive engineering has enabled single AAV packaging. This involves using compact promoters, minimizing redundant sequences, and selecting appropriately sized editors [26].
Non-Viral Delivery Methods
Lipid Nanoparticles (LNPs) have emerged as promising vehicles for prime editing component delivery [3]. LNPs can encapsulate both mRNA encoding the prime editor protein and pegRNA, protecting these molecules during transit and enabling efficient cellular uptake. Recent advances have improved LNP packaging capacity and cell-type specificity.
Electroporation effectively delivers prime editing ribonucleoprotein (RNP) complexes to ex vivo cells, including hematopoietic stem cells and T-cells [3]. This approach provides transient editor expression, potentially reducing off-target effects.
Polymer-based nanoparticles and other synthetic delivery systems offer additional non-viral options with potential for repeated administration and reduced immunogenicity compared to viral vectors.
Experimental Protocols for Delivery Optimization
Assessing Delivery Efficiency
Evaluating prime editing delivery success requires multiple complementary methods:
ddPCR and Next-Generation Sequencing: Droplet digital PCR provides highly quantitative measurements of editing efficiency, while NGS enables comprehensive characterization of editing outcomes and indel profiles [50]. These methods should target both the genomic locus of interest and, when using viral delivery, vector persistence.
Fluorescent Reporter Systems: Engineered fluorescent reporter cells enable rapid assessment of editing efficiency through flow cytometry or microscopy [50]. These systems typically place a target sequence with a premature stop codon before a fluorescent protein, where successful editing restores expression.
T7 Endonuclease I (T7EI) Assay: This mismatch detection method provides semi-quantitative editing assessment through gel electrophoresis, though it lacks the sensitivity of more advanced techniques [50].
pegRNA Design and Validation Protocol
Optimal pegRNA design follows this established workflow:
Target Site Selection: Identify potential target sites with PAM sequences (NGG for SpCas9) appropriately positioned relative to the edit location.
pegRNA Construction:
- Spacer sequence: 20 nucleotides complementary to target DNA
- PBS length: Test 10-15 nucleotides, starting with 13 nt
- RTT length: 10-16 nucleotides for most edits, extended for larger modifications
- Optimal GC content: 40-60% for PBS [49]
Stability Enhancements: Incorporate RNA pseudoknot motifs (epegRNA) or polyU tracts (for PE7) to protect against exonuclease degradation [9] [26].
Validation: Test multiple pegRNA designs with varying PBS and RTT lengths to identify optimal configurations for each target.
Mismatch Repair Manipulation
Cellular DNA mismatch repair (MMR) pathways can recognize and reverse prime edits, reducing efficiency. The PE4 and PE5 systems address this limitation by incorporating a dominant-negative MLH1 mutation (MLH1dn) to transiently inhibit MMR [15] [26]. This approach increases editing efficiency 2.0 to 7.7-fold across different cell types while minimizing unintended mutations [26].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Prime Editing Delivery Studies
| Reagent/Category | Function | Examples/Specifications |
|---|---|---|
| Prime Editor Plasmids | Editor expression | PE2, PEmax, PE4, PE6 variants |
| pegRNA Expression Systems | pegRNA delivery | U6-promoter vectors, synthetic pegRNAs |
| Viral Delivery Systems | In vivo delivery | AAV serotypes (AAV9 for broad tropism) |
| Non-Viral Delivery Agents | In vitro/ ex vivo delivery | Lipid nanoparticles, electroporation systems |
| Validation Tools | Efficiency assessment | ddPCR assays, NGS libraries, reporter cells |
| MMR Modulators | Enhance editing efficiency | MLH1dn expression constructs |
| pegRNA Stabilizers | Improve pegRNA longevity | epegRNA scaffolds, La fusion constructs |
Prime editing represents one of the most precise genome editing technologies available, yet its substantial size has necessitated innovative delivery solutions. Current strategies including split systems, compact editors, and advanced delivery vehicles have demonstrated promising results in model systems. The ongoing development of even smaller, more efficient prime editors, combined with improved delivery platforms, will likely overcome current limitations. As these technologies mature, prime editing is poised to enable new therapeutic strategies for genetic diseases that have previously been intractable to gene correction approaches. The field continues to evolve rapidly, with each advance bringing us closer to realizing the full therapeutic potential of precision genome editing.
Prime editing represents a significant leap forward in precision genome editing, enabling the programmable installation of precise base substitutions, insertions, and deletions without inducing double-strand DNA breaks (DSBs) or requiring donor DNA templates [9] [16]. This "search-and-replace" technology utilizes a fusion protein consisting of a Cas9 nickase (H840A) and an engineered reverse transcriptase (RT), programmed by a specialized prime editing guide RNA (pegRNA) that both specifies the target site and encodes the desired edit [1] [3]. Despite its revolutionary potential, initial prime editor systems suffered from limitations, particularly low and variable editing efficiencies that hindered robust application across diverse cell types and genetic targets [17] [15]. These challenges have driven the development of engineered solutions, primarily focusing on two key areas: the stabilization and optimization of pegRNAs through engineered structures (epegRNAs) and systematic enhancement of the editor protein itself and its cellular environment. This whitepaper examines these critical engineering advancements, providing a technical guide for researchers and drug development professionals seeking to implement state-of-the-art prime editing technologies in their work.
Engineering and Optimizing pegRNAs
Structural Limitations of Original pegRNAs
The original pegRNAs faced a fundamental stability issue: their single-stranded 3' extensions were prone to degradation by cellular exonucleases, significantly reducing the amount of intact pegRNA available for successful editing [9]. This degradation directly limited the efficiency of prime editing, as the reverse transcriptase template (RTT) and primer binding site (PBS) — both located in the 3' extension — are essential for specifying and installing edits [49]. Furthermore, the extended length of pegRNAs (typically 120-145 nucleotides) compared to standard sgRNAs presented challenges for synthesis, delivery, and cellular stability [3]. These limitations necessitated engineering approaches to protect pegRNA integrity and enhance their functional persistence within cells.
Engineered pegRNAs (epegRNAs) and Their Protective Motifs
The development of engineered pegRNAs (epegRNAs) addressed pegRNA instability by incorporating structured RNA motifs at the 3' end, which act as protective elements against exonuclease degradation [9] [15]. These motifs significantly improve editing efficiency by increasing the half-life of the pegRNA in cells, ensuring that more editor complexes are programmed with intact templates for reverse transcription. Different structural motifs have been successfully employed, offering researchers multiple options for optimization:
- tevopreQ1 Motif: This structured RNA motif, derived from a bacterial preQ1 riboswitch, was among the first protective elements validated for epegRNAs. Its incorporation typically improves prime editing efficiency by approximately 3-4 fold across multiple human cell lines and primary human fibroblasts without increasing off-target effects [51] [17].
- mpknot and evopreQ1 Motifs: These alternative structured RNA motifs function similarly to tevopreQ1 by creating stable tertiary structures that physically block exonuclease access to the 3' end of the pegRNA [9].
- Zika Virus Exoribonuclease-Resistant RNA Motif (xr-pegRNA): This viral-derived motif provides exceptional resistance to specific exonuclease activities, further enhancing pegRNA stability in challenging cellular environments [9].
- G-Quadruplex (G-PE): G-rich sequences that form stable four-stranded structures offer an alternative stabilization strategy, particularly effective in certain genomic contexts [9].
Table 1: Comparison of epegRNA Protective Motifs
| Motif Type | Typical Efficiency Gain | Key Characteristics | Considerations |
|---|---|---|---|
| tevopreQ1 | 3-4 fold | Well-characterized, reliable performance | Standard choice for most applications |
| mpknot | 2-3 fold | Stable tertiary structure | May require linker optimization |
| xr-pegRNA | 3-5 fold | Exceptional exonuclease resistance | Viral-derived sequence |
| G-Quadruplex | 2-4 fold | Sequence-dependent formation | May have context-specific effects |
Critical Design Parameters for pegRNAs
Beyond protective motifs, several key parameters significantly impact pegRNA performance. Optimal design requires careful consideration of these factors:
- Primer Binding Site (PBS) Optimization: The PBS should be tested at different lengths, typically starting with 13 nucleotides, and should exhibit 40-60% GC content for optimal performance [49]. The PBS must be complementary to the DNA sequence immediately adjacent to the nick site to properly prime reverse transcription.
- Reverse Transcriptase Template (RTT) Design: The RTT should encode the desired edit along with sufficient downstream homology (typically 10-16 nucleotides) to facilitate flap equilibration and recombination [49]. For longer edits, testing different RTT lengths is crucial to avoid unfavorable secondary structures.
- 3' End Sequence Considerations: The first base of the pegRNA's 3' extension should not be cytosine (C), as this base may pair with G81 of the gRNA scaffold, disrupting canonical structure and Cas9 binding [49].
- Strategic Edit Design: Whenever possible, edits should be designed to disrupt the protospacer adjacent motif (PAM) to prevent re-binding and re-nicking of the newly edited strand, which can lead to indels and other byproducts [49]. Additionally, creating "bubbles" of 3 or more mismatched bases (by including silent mutations alongside primary edits) can help evade cellular mismatch repair (MMR) systems that might otherwise revert edits [49].
Diagram: epegRNA Structural Components and Design Parameters
System-Level Enhancements for Improved Prime Editing
Evolution of Prime Editor Architectures
The prime editing system has evolved through multiple generations, each introducing significant improvements to editing efficiency and precision:
- PE1 to PE2: The original PE1 system demonstrated proof-of-concept but showed limited efficiency. PE2 incorporated an engineered reverse transcriptase (with mutations H9Y, D200N, T306K, W313F, T330P, and L603W) that significantly improved the affinity and stability of RNA-DNA substrates, resulting in enhanced editing efficiency [1] [16].
- PE3 and PE3b Systems: PE3 further increased efficiency by incorporating an additional sgRNA that nicks the non-edited strand, encouraging the cell to use the edited strand as a repair template [1] [15]. PE3b represents a refined approach where the nicking sgRNA is designed to bind only after the edit is installed, reducing concurrent nicks and minimizing indel formation [49].
- PEmax Architecture: This optimized editor system improves nuclear localization and expression through codon optimization and the addition of nuclear localization signals, enhancing performance across diverse cell types [17] [15].
Table 2: Evolution of Prime Editing Systems
| System | Key Components | Editing Efficiency | Key Advantages | Limitations |
|---|---|---|---|---|
| PE1 | Cas9(H840A)-WT RT + pegRNA | ~10-20% in HEK293T [1] | Proof of concept | Low efficiency |
| PE2 | Cas9(H840A)-engineered RT + pegRNA | ~20-40% in HEK293T [1] | Improved RT efficiency | Still limited by MMR |
| PE3 | PE2 + nicking sgRNA | ~30-50% in HEK293T [1] | Enhanced efficiency via strand correction | Increased indel formation |
| PE3b | PE2 + edit-dependent nicking sgRNA | Similar to PE3 [15] | Reduced indels via specific nicking | Requires appropriate PAM |
| PEmax | Optimized PE2 + improved expression | 3-72× over PE2 [15] | Enhanced nuclear localization, expression | Larger coding sequence |
Manipulating Cellular DNA Repair Environments
A critical advancement in prime editing came from understanding and manipulating cellular DNA repair pathways, particularly the mismatch repair (MMR) system:
- MMR Inhibition (PE4 and PE5 Systems): PE4 and PE5 systems co-express a dominant-negative variant of the MLH1 protein (MLH1dn) to transiently inhibit MMR, which otherwise tends to revert prime edits [1] [15]. This approach can increase editing efficiencies by 2.5 to 5.5-fold in HEK293T cells, with particularly strong benefits for substitutions that are especially susceptible to MMR [15].
- MMR Evasion Through Edit Design: An alternative to direct MMR inhibition involves designing edits that naturally evade MMR recognition. Creating "bubbles" of 3 or more mismatched bases (by incorporating additional silent mutations) makes the heteroduplex less recognizable to MMR machinery, significantly improving editing persistence [49].
Specialized Systems for Therapeutic Applications
Recent engineering efforts have addressed specific challenges for therapeutic applications:
- Split Prime Editing (sPE) Systems: To overcome adeno-associated virus (AAV) delivery size constraints, sPE systems separate the nCas9 and RT components, allowing them to function independently while maintaining editing capability [9]. This innovation enables dual-AAV delivery for in vivo applications.
- PE7 System with La Protein Fusion: This system fuses the prime editor with the human La protein, an endogenous RNA-binding protein that stabilizes RNAs with 3' polyU tracts [49]. Adding 3' polyU tracts to pegRNAs (but not epegRNAs) enhances their stability through La binding, improving editing outcomes in challenging cell types [51] [49].
Computational Design and High-Throughput Screening
AI and Machine Learning Approaches
Artificial intelligence (AI) and machine learning are increasingly applied to optimize prime editing systems [51]. These computational approaches accelerate the development process by:
- Predicting highly active pegRNA designs based on sequence features and structural accessibility [51]
- Guiding protein engineering of improved editor variants through deep mutational scanning and evolutionary analysis [51]
- Supporting the discovery of novel genome-editing enzymes from microbial systems by identifying patterns in protein sequences and structures [51]
- Enabling the development of predictive models for editing outcomes based on cellular context and target sequence features [51]
Tools such as the pegRNA Linker Identification Tool (pegLIT) help design optimal linkers for epegRNAs with large structured motifs like mpknot, minimizing unwanted intra-RNA base pairing that could compromise function [49].
Benchmarking and Validation Platforms
Rigorous benchmarking is essential for validating prime editing performance. Recent work has established high-efficiency prime editing platforms capable of multiplexed dropout screening, using libraries of ~240,000 epegRNAs targeting ~17,000 codons [17]. These platforms enable:
- Functional interrogation of small genetic variants at scale
- Identification of negative selection phenotypes for nonsense mutations targeted to essential genes
- Evaluation of synonymous mutations that disrupt splice site motifs
- Assessment of editing specificity through codon-matched controls
Such systematic approaches provide robust datasets for training AI models and establishing design principles that generalize across genomic contexts.
Experimental Protocols and Methodologies
Implementing High-Efficiency Prime Editing
For researchers establishing prime editing capabilities, the following protocol outlines key steps for achieving high-efficiency editing:
Cell Line Engineering and Culture Conditions
- Establish clonal cell lines (e.g., K562) that constitutively express optimized prime editor proteins (PE2, PEmax) with fluorescent markers (e.g., EGFP) to monitor editor expression [17].
- For MMR-deficient systems, genetically disrupt MLH1 in PEmax-derived cell lines (creating PEmaxKO lines) to enhance editing efficiency, particularly for substitutions susceptible to MMR [17].
- Maintain editor expression stability through appropriate antibiotic selection and regular monitoring of fluorescent markers.
pegRNA Delivery and Editing Timelines
- Transduce cells with pegRNAs or epegRNAs at low multiplicity of infection (MOI of ~0.7) to minimize multiple integrations [17].
- Select for successfully transduced cells using appropriate antibiotic resistance markers.
- Allow extended editing timelines (up to 28 days) for accumulation of precise edits, with sampling at 7-day intervals to monitor progression [17].
- For transient delivery, utilize optimized protocols for nucleofection or lipofection of pegRNA plasmids or ribonucleoprotein (RNP) complexes.
Evaluation and Validation of Editing Outcomes
- Sequence target loci using high-throughput amplicon sequencing to comprehensively characterize editing outcomes.
- Quantify three key outcome categories: (1) precise edits only, (2) edits with at least one error, and (3) unedited sequence [17].
- Assess editing specificity by analyzing potential off-target sites with sequence similarity to the pegRNA spacer, PBS, and RTT regions.
- For therapeutic applications, perform whole-genome sequencing to exclude guide RNA-independent off-target mutations [52].
Diagram: Prime Editing Experimental Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Prime Editing Experiments
| Reagent Category | Specific Examples | Function and Application | Considerations |
|---|---|---|---|
| Editor Plasmids | pCMV-PE2, PEmax constructs | Express the prime editor fusion protein | PEmax offers improved nuclear localization and expression |
| pegRNA Backbones | tevopreQ1, mpknot vectors | Templates for synthesizing pegRNAs with protective motifs | Choice of motif affects stability and efficiency |
| Cell Lines | HEK293T, K562, PEmaxKO (MLH1-) | Provide cellular context for editing | MMR-deficient lines enhance certain edits |
| Delivery Tools | Lentiviral systems, nucleofection reagents | Introduce editing components into cells | Consider size constraints for viral delivery |
| MMR Inhibitors | MLH1dn expression constructs | Temporarily suppress mismatch repair | Particularly useful for single-base substitutions |
| Selection Markers | Puromycin, blasticidin resistance | Enrich for successfully transduced cells | Concentration requires cell-type optimization |
| Screening Libraries | Self-targeting sensor libraries [17] | Benchmark editing efficiency across many targets | Enable multiplexed assessment of pegRNA performance |
The strategic engineering of epegRNAs and system-level enhancements has dramatically advanced prime editing technology from a proof-of-concept to a robust, efficient platform for precision genome manipulation. The combination of protective RNA motifs, optimized editor architectures, and cellular environment manipulation has enabled editing efficiencies exceeding 90% for many targets in validated systems [17]. These improvements have opened new avenues for research and therapeutic development, including multiplexed functional genomics screens, disease modeling, and correction of pathogenic mutations in therapeutic contexts.
Future developments will likely focus on further enhancing delivery efficiency through continued miniaturization of editing components, improving cell-type specificity through advanced targeting approaches, and refining predictive algorithms for editing outcomes across diverse genomic contexts. As these engineering solutions continue to mature, prime editing is poised to become an increasingly indispensable tool for researchers and drug development professionals pursuing precise genetic interventions.
Validation and Comparative Analysis: Positioning Prime Editing in the Genome Editing Landscape
The advent of CRISPR-Cas9 technology revolutionized genetic engineering by providing researchers with an accessible, programmable system for modifying genomes. However, its reliance on creating double-strand breaks (DSBs) in DNA has inherent limitations and safety concerns, including unwanted mutations and chromosomal rearrangements. Prime editing represents a significant technological advancement that addresses many of these limitations by enabling precise genetic modifications without requiring DSBs. This whitepaper provides an in-depth technical comparison of these two genome-editing platforms, focusing on their mechanisms, safety profiles, and precision for research and therapeutic applications. Understanding the relative advantages and limitations of each system is crucial for researchers and drug development professionals selecting the appropriate tool for specific genetic modifications.
Fundamental Mechanisms: How CRISPR-Cas9 and Prime Editing Work
CRISPR-Cas9: A Double-Strand Break-Based System
The CRISPR-Cas9 system operates through a relatively simple mechanism involving the Cas9 nuclease and a guide RNA (gRNA). The gRNA directs Cas9 to a specific DNA sequence, where the enzyme creates a double-strand break (DSB) [53]. This break activates the cell's innate DNA repair mechanisms, primarily non-homologous end joining (NHEJ) or homology-directed repair (HDR). NHEJ often results in small insertions or deletions (indels) that can disrupt gene function, making it useful for gene knockout studies. HDR can incorporate a donor DNA template to achieve precise changes but operates at significantly lower efficiency than NHEJ in most cell types [54]. The DSB-dependent nature of CRISPR-Cas9 is both its greatest strength and most significant limitation, as these breaks can lead to unintended genomic consequences.
Prime Editing: A "Search-and-Replace" System Without Double-Strand Breaks
Prime editing represents a paradigm shift in precision genome editing. Rather than creating DSBs, it uses a fusion protein consisting of a Cas9 nickase (H840A) and a reverse transcriptase (RT), programmed with a specialized prime editing guide RNA (pegRNA) [9] [48]. The pegRNA not only directs the complex to the target DNA site but also contains a template for the desired edit. The system works through a multi-step process: first, the Cas9 nickase nicks one DNA strand, creating a priming site; second, the reverse transcriptase uses the pegRNA template to synthesize DNA containing the desired edit; finally, cellular repair mechanisms incorporate this edit into the genome [3] [55]. This "search-and-replace" capability allows prime editing to achieve all 12 possible base-to-base conversions, small insertions, and deletions without DSBs or donor DNA templates [1].
Table 1: Core Components of CRISPR-Cas9 and Prime Editing Systems
| Component | CRISPR-Cas9 | Prime Editing |
|---|---|---|
| Core Enzyme | Cas9 nuclease | Cas9 nickase (H840A) fused to reverse transcriptase |
| Guide RNA | Single guide RNA (sgRNA, ~100 nt) | Prime editing guide RNA (pegRNA, ~120-145 nt) |
| Template Requirement | Donor DNA needed for precise edits | pegRNA contains edit template; no donor DNA needed |
| DNA Cleavage | Double-strand break | Single-strand nick |
| Key Innovations | Programmable DNA cleavage | Reverse transcription at target site |
Visualizing the Core Mechanisms
The diagram below illustrates the fundamental mechanistic differences between CRISPR-Cas9 and prime editing systems, highlighting their distinct approaches to genome modification.
Safety and Precision Profiles: A Comparative Analysis
Genomic Safety and Unintended Effects
The safety profiles of CRISPR-Cas9 and prime editing differ substantially due to their distinct mechanisms of action. CRISPR-Cas9's reliance on DSBs presents several well-documented safety concerns. Beyond small indels at the target site, CRISPR-Cas9 can cause large structural variations (SVs), including chromosomal translocations and megabase-scale deletions [53]. These SVs raise substantial safety concerns for clinical applications, as they can disrupt multiple genes or regulatory regions. Additionally, off-target editing at genomic sites with sequence similarity to the intended target can occur, though high-fidelity Cas9 variants have reduced this risk [53].
Prime editing demonstrates a superior safety profile in several key aspects. By avoiding DSBs, it significantly reduces the risk of large SVs and chromosomal abnormalities [9] [48]. The system also shows reduced off-target effects compared to standard CRISPR-Cas9, as the nickase activity is inherently less prone to off-target modifications than DSB creation [1]. However, prime editing is not completely risk-free. Recent studies indicate that nick-based platforms may still generate unintended genetic alterations, including small indels, though at much lower frequencies than CRISPR-Cas9 [53].
Table 2: Safety Profile Comparison of CRISPR-Cas9 and Prime Editing
| Safety Parameter | CRISPR-Cas9 | Prime Editing |
|---|---|---|
| Double-Strand Breaks | Yes, required for activity | No, uses single-strand nicks |
| Large Structural Variations | Significant risk of kilobase-to megabase-scale deletions and translocations [53] | Greatly reduced risk |
| Off-Target Editing | Moderate to high risk, improved with high-fidelity variants | Lower risk due to nicking mechanism |
| On-Target Indels | Frequent | Infrequent |
| Bystander Editing | Not applicable | Minimal compared to base editors |
| Impact of DNA Repair Inhibition | Can exacerbate genomic aberrations [53] | Less dramatic impact |
Editing Precision and Versatility
When comparing precision and editing capabilities, prime editing offers significant advantages in versatility while having different efficiency constraints. CRISPR-Cas9 is highly efficient at generating gene knockouts through indel formation but is relatively inefficient at achieving precise nucleotide changes, typically requiring HDR with donor DNA templates. HDR efficiency varies considerably by cell type and cell cycle stage, with post-mitotic cells being particularly challenging targets.
Prime editing expands the scope of precise genome editing considerably. It can achieve all 12 possible base-to-base conversions, while base editors (another CRISPR-derived technology) are limited primarily to C-to-T and A-to-G transitions [9] [1]. Prime editing also enables targeted insertions and deletions without DSBs. However, editing efficiency is highly variable across genomic loci and cell types, with some targets proving more challenging than others [48] [55]. The system has undergone rapid evolution since its initial development, with PE2, PE3, PEmax, and PE6 variants progressively improving editing efficiency through optimization of the reverse transcriptase and Cas9 components [48] [1] [35].
Table 3: Editing Capabilities and Performance Comparison
| Editing Parameter | CRISPR-Cas9 | Prime Editing |
|---|---|---|
| Base Substitutions | Limited (requires HDR) | All 12 possible conversions [1] |
| Insertions | Possible with HDR (limited size) | Small insertions (typically <几十bp) |
| Deletions | Efficient via NHEJ | Small, precise deletions |
| Efficiency (Precise Edits) | Low for HDR (typically 0.1-20%) | Variable by locus (1-50%+ with optimized systems) [1] |
| Efficiency (Gene Knockout) | High (often >70%) | Not designed for knockouts |
| PAM Requirement | NGG (for SpCas9) | NGG (for SpCas9) |
| Dependence on Cell Division | Required for HDR | Works in both dividing and non-dividing cells [48] |
| Therapeutic Applications | Approved (Casgevy for SCD/TDT) [14] | First clinical trials underway (PM359 for CGD) [35] |
Technical Considerations for Research Applications
Experimental Design and Workflow
Implementing prime editing requires careful experimental design distinct from CRISPR-Cas9 workflows. A critical first step is pegRNA design, which involves not only the spacer sequence that targets the editor to the desired genomic locus but also the primer binding site (PBS) and reverse transcription template (RTT) containing the desired edit [3]. The length and melting temperature of the PBS significantly impact editing efficiency, with optimal PBS characteristics varying by cell type and target locus [55]. Numerous computational tools have been developed to assist with pegRNA design, incorporating factors such as secondary structure and potential off-target binding.
For CRISPR-Cas9, the workflow is more straightforward, involving design of a standard sgRNA with high on-target efficiency and minimal off-target potential. For precise editing requiring HDR, a donor DNA template must be designed with homology arms flanking the edit, typically extending 400-800 base pairs on each side for most mammalian cell applications.
Delivery Considerations
Both systems face challenges in delivery, though prime editing presents additional complexities due to the larger size of its components. The prime editor fusion protein is substantially larger than standard Cas9, and the pegRNA is longer than conventional sgRNAs, creating challenges for packaging into delivery vectors with limited capacity, particularly adeno-associated viruses (AAVs) [3] [35]. Creative solutions have emerged, including the use of dual AAV systems and the development of compact prime editors such as PE6b, which is approximately 33% smaller than PEmax while maintaining comparable editing efficiency [48].
Lipid nanoparticles (LNPs) have emerged as a promising delivery vehicle for both systems, particularly for in vivo applications. LNPs have been successfully used to deliver mRNA encoding CRISPR-Cas9 components and have shown promise for prime editing delivery, as demonstrated in recent preclinical studies [14] [56].
Optimization Strategies
Maximizing editing efficiency for both systems often requires optimization. For prime editing, this can include:
- Testing multiple pegRNA designs for each target
- Using engineered pegRNAs (epegRNAs) with structured RNA motifs to protect against degradation [9] [1]
- Employing PE3 or PE3b systems that include an additional nicking sgRNA to enhance editing efficiency [48] [3]
- Transiently modulating DNA repair pathways, such as using MMR inhibitors, to improve editing outcomes [48] [54]
For CRISPR-Cas9 HDR applications, optimization strategies include:
- Synchronizing cells in S/G2 phases of the cell cycle
- Using HDR enhancers such as small molecules that temporarily inhibit NHEJ components
- Employing Cas9 fusion proteins that recruit HDR factors to the target site
Research Reagent Solutions and Experimental Tools
Successful implementation of genome editing technologies requires appropriate reagents and tools. The table below outlines key solutions for both CRISPR-Cas9 and prime editing systems.
Table 4: Essential Research Reagents and Tools for Genome Editing
| Reagent/Tool | Function | CRISPR-Cas9 | Prime Editing |
|---|---|---|---|
| Editor Expression Plasmid | Encodes the editor protein | Cas9 nuclease | PE2, PEmax, PE6 variants [48] [1] |
| Guide RNA Vector | Encodes sgRNA or pegRNA | U6-promoter driven sgRNA | U6-promoter driven pegRNA |
| Delivery Vehicles | Introduces editors into cells | LNPs, AAV, electroporation | Dual AAV systems, LNPs [35] |
| pegRNA Design Tools | Optimizes pegRNA design | Not applicable | Computational tools for PBS/RTT design |
| MMR Inhibitors | Enhances editing efficiency | Not typically used | MLH1dn improves prime editing efficiency [48] |
| Validation Assays | Confirms edits | Sanger sequencing, NGS | Specialized amplicon sequencing for precise edits |
| Off-Target Assessment | Detects unintended edits | GUIDE-seq, CIRCLE-seq | CHANGE-seq, specialized for PE |
The comparison between prime editing and CRISPR-Cas9 reveals a fundamental trade-off between precision and efficiency in current genome editing technologies. CRISPR-Cas9 remains the superior choice for applications where gene disruption is desired, offering high efficiency and well-established protocols. However, for precise nucleotide changes, small insertions, or deletions, prime editing provides a safer, more versatile alternative that avoids the pitfalls of double-strand break repair.
As prime editing technology continues to evolve, with ongoing improvements in efficiency, specificity, and delivery, its application in both basic research and therapeutic development is expected to expand rapidly. The recent clearance of the first prime editing clinical trial (PM359 for chronic granulomatous disease) marks a significant milestone in the translation of this technology [35]. For research teams, the choice between these platforms should be guided by the specific genetic modification required, the cellular context, and the relative importance of efficiency versus precision for the application at hand. Both systems will undoubtedly continue to play crucial roles in the genome editing toolkit, enabling researchers to address increasingly complex biological questions and develop innovative genetic therapies.
The advent of precision genome editing has revolutionized genetic research and therapeutic development. Among the most significant advancements are base editing and prime editing, two technologies that enable precise DNA alterations without relying on double-strand breaks (DSBs). This whitepaper provides a comprehensive technical comparison of these systems, examining their molecular mechanisms, editing scopes, efficiencies, and applications within biomedical research. We detail experimental protocols for implementing each technology and provide a structured analysis of their respective advantages and limitations, offering researchers a framework for selecting appropriate editing tools for specific experimental or therapeutic objectives.
Precision genome editing represents a pivotal advancement in genetic engineering, enabling targeted modifications with unprecedented accuracy. Traditional CRISPR-Cas9 systems, while revolutionary, operate by inducing double-strand breaks (DSBs) in DNA, which can lead to unintended insertions, deletions (indels), and chromosomal rearrangements [1] [2]. To overcome these limitations, two innovative technologies have emerged: base editing and prime editing.
Base editing, developed in 2016, utilizes a catalytically impaired Cas protein fused to a deaminase enzyme to directly convert one DNA base into another without causing DSBs [57] [58]. Prime editing, introduced in 2019, represents a more versatile "search-and-replace" system that couples a Cas9 nickase with a reverse transcriptase, programmed by a specialized guide RNA to install precise edits [1] [4]. Both technologies have rapidly evolved through multiple generations of optimization, expanding their capabilities and efficiencies.
This whitepaper examines the technical specifications, scope, and flexibility of base editing and prime editing systems, providing researchers with a comprehensive guide for their application in functional genomics, disease modeling, and therapeutic development.
Base Editing Architecture and Mechanism
Base editors are fusion proteins that combine a catalytically impaired Cas protein (either dead Cas9/dCas9 or nickase Cas9/nCas9) with a nucleoside deaminase enzyme [57] [59]. The system functions without creating DSBs, instead chemically modifying bases within a defined "editing window" of single-stranded DNA exposed during R-loop formation.
Table: Base Editor Components and Functions
| Component | Type/Variant | Function |
|---|---|---|
| Cas Protein | dCas9 (catalytically dead) | DNA binding without cleavage |
| nCas9 (nickase) | Binds DNA and nicks non-edited strand | |
| Deaminase Enzyme | Cytidine deaminase (e.g., APOBEC1) | Converts cytosine (C) to uracil (U) |
| Adenine deaminase (e.g., engineered TadA) | Converts adenine (A) to inosine (I) | |
| Accessory Proteins | Uracil glycosylase inhibitor (UGI) | Prevents repair of C→U conversion |
| Guide RNA | Standard sgRNA | Targets complex to specific genomic locus |
The base editing process involves three key steps: (1) the gRNA directs the base editor to the target DNA sequence, forming an R-loop that exposes a single-stranded DNA region; (2) the deaminase enzyme modifies specific bases within the editing window (typically nucleotides 4-8 in the protospacer); and (3) cellular repair mechanisms or DNA replication permanent the edits [58] [59].
Two primary base editor classes have been developed:
- Cytosine Base Editors (CBEs): Convert C•G to T•A base pairs using cytidine deaminase enzymes. The original BE3 system consisted of nCas9 fused to rat APOBEC1 and UGI, achieving C→T conversion with reduced indel formation compared to CRISPR-Cas9 [58].
- Adenine Base Editors (ABEs): Convert A•T to G•C base pairs using engineered tRNA adenosine deaminase (TadA). The first highly efficient ABE (ABE7.10) was developed through extensive protein evolution to create a DNA-compatible adenine deaminase [58] [59].
Prime Editing Architecture and Mechanism
Prime editing represents a more versatile genome editing platform that enables precise insertions, deletions, and all 12 possible base-to-base conversions without requiring DSBs or donor DNA templates [1] [4]. The system consists of two primary components:
- Prime Editor Protein: A fusion of Cas9 nickase (H840A) with an engineered reverse transcriptase (RT) from the Moloney murine leukemia virus (MMLV-RT)
- Prime Editing Guide RNA (pegRNA): A specialized RNA that both specifies the target site and encodes the desired edit
Table: Prime Editor Evolution and Characteristics
| Generation | Components | Editing Efficiency | Key Features |
|---|---|---|---|
| PE1 | nCas9 (H840A) + M-MLV RT | ~10-20% | Initial proof-of-concept |
| PE2 | Optimized RT + nCas9 | ~20-40% | Enhanced efficiency and fidelity |
| PE3 | PE2 + additional sgRNA | ~30-50% | Dual nicking strategy increases efficiency |
| PE4/PE5 | PE3 + MLH1dn | ~50-80% | MMR inhibition enhances editing |
| PE6 | Compact RT variants + epegRNAs | ~70-90% | Improved delivery and pegRNA stability |
| PE7 | La protein fusion | ~80-95% | Enhanced pegRNA stability in challenging cells |
The prime editing mechanism involves five key steps [1] [4] [3]:
- Target binding: The PE-pegRNA complex binds to the target DNA sequence
- DNA nicking: The nCas9 domain nicks the non-target DNA strand
- Reverse transcription: The exposed 3' end hybridizes with the primer binding site (PBS) of the pegRNA, and the RT synthesizes DNA containing the desired edit using the RT template
- Flap resolution: Cellular enzymes mediate flap exchange, incorporating the edited strand
- DNA repair: Mismatch repair fixes the complementary strand to match the edit
The more recent PE3 system incorporates an additional sgRNA that nicks the non-edited strand to bias cellular repair toward the edited strand, increasing efficiency approximately 4.2-fold over PE2 [1] [4].
Comparative Analysis: Scope and Flexibility
Editing Scope and Capabilities
The fundamental distinction between base editing and prime editing lies in their scope of editable modifications. Base editors facilitate specific transition mutations, while prime editors support a broader range of precise genetic changes.
Table: Editing Capabilities Comparison
| Edit Type | Base Editing | Prime Editing |
|---|---|---|
| Transition Mutations | C→T, G→A, A→G, T→C | All possible (C→T, G→A, A→G, T→C, C→A, C→G, G→C, G→T, A→C, A→T, T→A, T→G) |
| Transversion Mutations | Not supported | All possible |
| Small Insertions | Not supported | Up to dozens of base pairs |
| Small Deletions | Not supported | Up to hundreds of base pairs |
| Combination Edits | Not supported | Yes (e.g., simultaneous substitution + insertion) |
Base editors are limited to four specific transition mutations: C→T and G→A (using CBEs) or A→G and T→C (using ABEs) [57] [58]. This restriction arises from the specific chemistry of deaminase enzymes, which cannot mediate transversion mutations (e.g., C→A, C→G, A→C, A→T, etc.).
In contrast, prime editing theoretically supports all 12 possible base-to-base conversions, in addition to targeted insertions (up to dozens of base pairs) and deletions (up to hundreds of base pairs) [1] [3]. This versatility stems from the template-directed nature of reverse transcription, which can encode any desired DNA sequence within the RT template region of the pegRNA.
Technical Specifications and Performance
Table: Technical Specifications Comparison
| Parameter | Base Editing | Prime Editing |
|---|---|---|
| DSB Formation | Minimal (nickase-based) | Minimal (nickase-based) |
| Typical Efficiency | 50-80% (optimized systems) | 20-50% (PE3), up to 90% with latest systems |
| Editing Window | Narrow (typically 4-5 nucleotides) | Flexible (determined by pegRNA design) |
| PAM Requirement | Yes (NGG for SpCas9) | Yes (NGG for SpCas9) |
| Bystander Edits | Common (multiple bases in window) | Rare (precise targeting) |
| Indel Formation | Low (0.1-1.0%) | Very low (<0.1% with optimized systems) |
| Off-Target Effects | DNA and RNA off-target editing observed | Minimal reported off-target activity |
Base editors typically achieve higher editing efficiencies (50-80% in optimized systems) but produce bystander edits when multiple editable bases fall within the editing window [58] [59]. The confined editing window (typically nucleotides 4-8 in the protospacer for SpCas9-based editors) restricts targeting flexibility and requires precise positioning relative to the PAM sequence.
Prime editing efficiencies vary considerably based on target locus, cell type, and pegRNA design, with early systems achieving 20-50% efficiency in human cells [1] [4]. Recent advancements like PE4/PE5 (incorporating MMR inhibitors) and PE6/PE7 (featuring optimized RTs and pegRNA stabilization) have boosted efficiencies to 70-95% for many targets [1]. Prime editing's key advantage lies in its precision, with minimal bystander edits or indel formation compared to base editing.
PAM Constraints and Targeting Scope
Both technologies face targeting constraints imposed by protospacer adjacent motif (PAM) requirements of their Cas9 components. The canonical SpCas9 requires a 5'-NGG-3' PAM sequence immediately downstream of the target site, restricting editable genomic loci [60].
Base editing has more stringent positioning requirements, as the target base must fall within the narrow editing window (typically 4-8 nucleotides upstream of the PAM) [60] [58]. This constraint limits the proportion of pathogenic SNPs targetable by base editors, estimated at approximately 30% for C→T corrections and 25% for A→G corrections [60].
Prime editing offers greater flexibility, as edits can be installed at various positions relative to the nick site (up to 10-15 nucleotides away) through pegRNA engineering [1] [61]. Recent developments like proPE (prime editing with prolonged editing window) further expand this range by using a second non-cleaving sgRNA to position the RT template near the edit site, enabling efficient editing beyond the typical prime editing range [61].
Experimental Protocols
Base Editing Experimental Workflow
Protocol: Implementing Base Editing in mammalian Cells
Target Selection and gRNA Design
- Identify target base and verify it falls within the editing window (positions 4-8 for SpCas9-based editors)
- Design gRNA with standard tools (e.g., CRISPick, CHOPCHOP), ensuring optimal positioning relative to PAM
- Select appropriate base editor: CBE (e.g., BE4) for C→T edits, ABE (e.g., ABE8e) for A→G edits
Editor Delivery
- Prepare expression plasmid encoding base editor and gRNA, or assemble as RNP complex
- Deliver to cells via appropriate method:
- mammalian Cells*: Lipofection, electroporation, or viral transduction (lentiviral, AAV)
- In vivo: Lipid nanoparticles (LNPs) or AAV vectors
Editing Validation
- Harvest cells 48-72 hours post-delivery
- Extract genomic DNA and amplify target region by PCR
- Analyze editing efficiency by Sanger sequencing (decoupled using BEAT or similar tools) or next-generation sequencing
- Assess potential off-target effects through targeted sequencing of predicted off-target sites or whole-genome sequencing
Optimization
- Test multiple gRNAs with different spacer lengths if efficiency is low
- For CBEs, consider UGI co-expression if purity is suboptimal
- Adjust delivery parameters and editor expression levels to minimize indel formation
Prime Editing Experimental Workflow
Protocol: Implementing Prime Editing in mammalian Cells
pegRNA Design
- Design spacer sequence (20 nt) complementary to target site
- Define desired edit(s) in reverse transcriptase template (RTT, typically 10-15 nt)
- Design primer binding site (PBS, typically 8-15 nt) with Tm ≈ 30°C
- Consider engineered pegRNA (epegRNA) modifications to enhance stability
Editor Assembly and Delivery
- Clone pegRNA expression cassette into appropriate vector
- Co-deliver prime editor (PE2, PE3, or PE5) and pegRNA expression constructs
- For PE3/PE3b systems, include additional nicking sgRNA expression construct
- Delivery methods similar to base editing, noting larger size of prime editor constructs
Editing Validation
- Harvest cells 72-96 hours post-delivery (longer timeframe accounts for slower editing process)
- Extract genomic DNA and amplify target region
- Analyze editing efficiency by targeted next-generation sequencing
- Verify edit specificity and assess potential indel formation
Optimization
- Systematically test multiple pegRNAs with varying PBS lengths and RTT designs
- For low-efficiency targets, consider PE4/PE5 systems with MMR inhibition
- Evaluate PE3 vs PE2 configurations to balance efficiency and indel formation
- For difficult-to-edit loci, consider proPE system using separate engRNA and tpgRNA [61]
Research Reagent Solutions
Table: Essential Research Reagents for Precision Genome Editing
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Base Editor Plasmids | BE4max, ABE8e, Target-AID | Optimized CBE and ABE expression |
| Prime Editor Plasmids | PE2, PE3, PE5, PE6 variants | Prime editor protein expression |
| pegRNA Expression Systems | ppegRNA, epegRNA vectors | Stable pegRNA transcription with modified scaffolds |
| Delivery Tools | AAV vectors, LNPs, Electroporation systems | Efficient editor delivery to target cells |
| Editing Reporters | PEAR plasmid, SURVEYOR assays | Rapid assessment of editing efficiency |
| Analysis Tools | BEAT, CRISPResso2, primary | Computational analysis of editing outcomes |
Base editing and prime editing represent complementary approaches in the precision genome editing toolkit, each with distinct advantages and limitations. Base editors offer higher efficiency for specific transition mutations but are constrained by their limited editing scope and potential for bystander edits. Prime editors provide unprecedented versatility in installing diverse genetic modifications but face challenges with efficiency and complex pegRNA design.
The selection between these technologies depends on specific research requirements: base editing excels for efficient correction of specific pathogenic point mutations falling within accessible sequence contexts, while prime editing is preferable for installing complex edits, transversion mutations, or modifications in sequences with restrictive editing windows.
Ongoing developments in both fields continue to address current limitations. For base editing, these include reducing off-target activity, narrowing editing windows, and expanding PAM compatibility. For prime editing, current research focuses on improving efficiency through engineered RT domains, optimized pegRNA designs, and MMR inhibition strategies. As both technologies mature, they promise to expand the therapeutic potential of precision genome editing for treating genetic disorders and advancing biomedical research.
Prime editing represents a significant advancement in precision genome editing by enabling targeted modifications without introducing double-strand DNA breaks (DSBs). This foundational difference from earlier CRISPR-Cas9 nuclease systems substantially reduces the risk of unintended on-target mutations such as insertions and deletions (indels) and large chromosomal rearrangements [1] [52]. The system utilizes a catalytically impaired Cas9 nickase (H840A) fused to an engineered reverse transcriptase (RT) and is directed by a specialized prime editing guide RNA (pegRNA) that both specifies the target genomic locus and encodes the desired edit [1] [3]. While this architecture inherently minimizes certain classes of off-target effects, comprehensive analytical methods are essential to quantify and validate its specificity, particularly for therapeutic applications where precision is paramount [52] [62].
Research consistently demonstrates that prime editing exhibits a favorable off-target profile. A critical study using whole-genome sequencing (WGS) revealed that prolonged expression of the PE2 editor in human pluripotent stem cells (hPSCs) did not generate guide RNA-independent off-target mutations in the genome. This finding stands in contrast to base editors, which can exhibit deaminase-mediated, guide-independent off-target effects across the genome and transcriptome [52]. The specificity of prime editing is therefore a key advantage, though rigorous, standardized assessment remains crucial as the technology evolves.
Despite its precision, prime editing can potentially generate unintended edits through several mechanisms. A primary concern is pegRNA-dependent off-target editing, where the pegRNA binds to and nicks genomic sites with sequence similarity to the intended target, leading to mis-incorporation of the edit encoded in its reverse transcriptase template (RTT) [1]. A second significant source is the formation of unintended on-target byproducts, including small indels. These indels can arise from errant double-strand breaks or imperfect resolution of the DNA flap intermediates during the editing process [62]. Recent engineering efforts have focused on understanding and mitigating this particular source of error.
A key discovery is that the cellular mismatch repair (MMR) pathway plays a dual role. While it can correct the edited:non-edited heteroduplex DNA to favor the unedited strand—thereby reducing editing efficiency—it can also sometimes convert nicks into DSBs, which subsequently generate indels during repair [17] [62]. Furthermore, the prime editor complex can, in some cases, extend the edited DNA strand past the end of the pegRNA template and into the pegRNA scaffold sequence, leading to the incorporation of scaffold-derived sequences into the genome [62]. Understanding these mechanisms is the first step toward developing more precise editors and accurate assessment protocols.
Engineering Editors for Enhanced Fidelity
Recent protein engineering efforts have successfully addressed the challenge of indel byproducts. By introducing mutations into the Cas9 nickase that relax nick positioning (e.g., K848A and H982A), researchers have engineered next-generation prime editors like the precise Prime Editor (pPE) and vPE. These mutations promote the degradation of the competing 5' DNA flap, which is often the source of indel errors. This strategic destabilization biases the flap equilibrium toward the incorporation of the edited 3' flap, thereby enhancing editing efficiency while dramatically reducing indel formation [62].
The performance improvements are substantial. Compared to the PEmax editor, the pPE variant reduced indel errors by an average of 26-fold (ranging from 7.7 to 36-fold across six genomic loci) in a PE3 editing configuration (using both a pegRNA and a nicking sgRNA). This resulted in edit-to-indel ratios as high as 361:1 [62]. This breakthrough demonstrates that the specificity of prime editing is not a fixed property but can be systematically improved through rational protein design to minimize inherent error-prone side reactions.
Methodologies for Assessing Off-Target Effects
A comprehensive off-target assessment strategy employs multiple complementary techniques, each capable of detecting different classes of unintended edits.
Genome-Wide Analysis
Whole-genome sequencing (WGS) represents the most unbiased and comprehensive method for detecting off-target effects. This approach sequences the entire genome of edited cells to identify single-nucleotide variants (SNVs), indels, and structural variations that deviate from the unedited control genome. Its major advantage is that it is hypothesis-free and does not require prior knowledge of potential off-target sites [52]. As demonstrated in hPSCs, WGS can conclusively show that prolonged PE2 expression does not cause an elevated mutation rate, providing a high level of confidence for therapeutic development [52]. The primary limitation of WGS is its cost and the computational challenge of analyzing the massive datasets it generates.
Targeted and In Silico Analysis
In silico prediction tools coupled with targeted deep sequencing offer a more focused and cost-effective strategy. Tools like PRIDICT2.0 use machine learning models trained on vast prime editing datasets to predict the efficiency of a given pegRNA. While their primary design is for optimizing on-target editing, the features they analyze (e.g., sequence homology, GC content) are also relevant for predicting potential off-target sites [31]. Researchers can use these tools to generate a list of putative off-target sites, which are then amplified and deeply sequenced from edited cell populations. This method provides sensitive, quantitative data on editing events at pre-selected loci but is inherently limited to the sites chosen for analysis.
The table below summarizes the key methodologies used for off-target analysis.
Table 1: Key Methodologies for Off-Target Analysis in Prime Editing
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Whole-Genome Sequencing (WGS) [52] | High-throughput sequencing of the entire genome from edited and control cells. | Unbiased, comprehensive detection of all variant types across the genome. | High cost; computationally intensive; lower depth may miss low-frequency events. |
| Targeted Deep Sequencing [31] | Deep sequencing of PCR-amplified genomic regions (including in silico predicted off-target sites). | Highly sensitive and quantitative for specific loci; cost-effective. | Limited to pre-selected regions; potential to miss unpredicted off-target sites. |
| In Silico Prediction (e.g., PRIDICT2.0) [31] | Machine learning models predict pegRNA efficiency and potential off-target activity based on sequence features. | Fast, inexpensive, and informative for guide design and risk assessment. | Predictive only; accuracy depends on training data; may generate false positives/negatives. |
Experimental Protocol for a Comprehensive Off-Target Assessment
The following workflow outlines a robust protocol for profiling off-target effects, integrating both computational and empirical methods.
Cell Line Preparation and Editing:
- Generate a clonal cell line that constitutively expresses the prime editor (e.g., PEmax) to ensure stable, long-term editor expression [17]. A suitable control is a cell line expressing a catalytically dead editor.
- Transduce the cells with the specific pegRNA of interest using lentiviral vectors at a low multiplicity of infection (MOI ~0.7) to ensure single-copy integration and minimize pegRNA over-expression effects [17].
- Culture the transduced cells for at least 14-28 days to allow edits to accumulate and stabilize, sampling at multiple time points to track the dynamics of edit installation and byproduct formation [17].
In Silico Off-Target Prediction:
- Utilize multiple computational tools to generate a list of potential off-target sites. Input the pegRNA spacer sequence into algorithms like Cas-OFFinder to find genomic loci with sequence similarity, allowing for mismatches and bulges.
- Use a model like PRIDICT2.0 to score these potential off-target sites based on features known to influence prime editing efficiency (e.g., edit type, edit position, local sequence context) [31].
Empirical Analysis via Targeted Sequencing:
- Design PCR primers to amplify the top 50-100 predicted off-target loci from the in silico analysis, as well as the intended on-target site.
- Perform high-depth (e.g., >10,000x coverage) amplicon sequencing on the genomic DNA from the edited and control cell populations.
- Use computational pipelines (e.g., CRISPResso2) to align sequences and quantify the frequency of precise edits, imprecise edits, and indels at each locus.
Genome-Wide Validation via WGS:
- Subject genomic DNA from the edited cell population and an appropriate control (e.g., the parent cell line or a cell line expressing a dead editor) to WGS.
- Sequence to a high coverage (recommended >30x) using an Illumina platform.
- Analyze the data using a standardized variant calling pipeline (e.g., GATK) to identify single-nucleotide variants and indels that are significantly enriched in the edited sample compared to the control.
Quantitative Analysis of Editing Outcomes
Rigorous quantification of editing outcomes is essential for a complete specificity profile. This involves analyzing the composition of sequencing reads from the on-target site to distinguish between precise edits, imprecise edits (e.g., edits with additional errors), and pure indels.
Key Outcome Metrics and Reagents
The table below defines key outcome metrics and lists essential research reagents for conducting these analyses.
Table 2: Key Metrics and Reagents for Specificity Analysis
| Category | Item / Metric | Description / Function |
|---|---|---|
| Key Metrics | Precise Editing Efficiency | The percentage of sequencing reads containing only the intended edit(s) with no additional changes [17]. |
| Indel Frequency | The percentage of sequencing reads containing small insertions or deletions, often at the edit site or flap junction [62]. | |
| Edit:Indel Ratio | The ratio of precise edits to indels; a key indicator of editing cleanliness and fidelity (e.g., 543:1 for vPE) [62]. | |
| Error Rate (with errors) | The percentage of reads containing the intended edit but with additional, unwanted nucleotide changes [17]. | |
| Research Reagents | Prime Editor Plasmids | Plasmids for expressing PE2, PEmax, or next-gen editors (e.g., pPE, vPE). PEmax offers improved nuclear localization and codon optimization [17] [62]. |
| pegRNA Expression Vectors | Plasmids or viral vectors for expressing pegRNAs or engineered pegRNAs (epegRNAs) with stability-enhancing motifs [17]. | |
| MMR-Inhibiting Components | Plasmids expressing dominant-negative MLH1 (MLH1dn) used in the PE4/PE5 systems to temporarily inhibit MMR and boost editing efficiency [1] [17]. | |
| Nicking sgRNA | A standard sgRNA that directs the editor to nick the non-edited strand in PE3/PE5 systems to increase editing efficiency [1]. | |
| Validated Cell Lines | Clonal cell lines (e.g., K562, HEK293T) with stable, constitutive expression of the prime editor for screening and benchmarking [17]. |
Performance of Advanced Prime Editors
Quantitative data from high-efficiency editing platforms demonstrates the significant progress made in reducing off-target byproducts. The use of MMR-deficient cell lines (e.g., PEmaxKO) or co-expression of MLH1dn (PE4 approach) can dramatically increase precise editing efficiency, in some cases achieving over 95% for certain edits, while keeping errors low [17]. This suggests that suppressing MMR not only boosts efficiency but can also reduce a source of error generation.
The most striking improvements come from engineered editors. As shown in the performance table below, the vPE editor achieves an exceptionally high edit-to-indel ratio, making it one of the cleanest editors available.
Table 3: Performance Comparison of Prime Editor Systems
| Prime Editor System | Key Features | Typical Precise Editing Efficiency* | Edit:Indel Ratio* |
|---|---|---|---|
| PE2 [1] | Reverse transcriptase fused to Cas9 nickase (H840A); uses pegRNA. | ~20-40% | Baseline |
| PE3 [1] | PE2 system with an additional nicking sgRNA to enhance efficiency. | ~30-50% | Lower than PE2 (due to increased nicking) |
| PE4/PE5 [1] | PE2/PE3 system with co-expression of MLH1dn to suppress MMR. | ~50-70% | Improved over PE2/PE3 |
| PEmax [17] | Codon-optimized PE2 with improved nuclear localization signals. | Higher than PE2 | Similar to PE2 |
| pPE / vPE [62] | Engineered with nickase mutations (e.g., K848A, H982A) to destabilize 5' flap and reduce indels. | Comparable to PEmax | Up to 543:1 (up to 60-fold lower indels than PEmax) |
Note: Efficiency and ratios are highly dependent on the target locus and edit type. Values are representative ranges from literature [1] [62].
The assessment of off-target effects has evolved in tandem with prime editing technology itself. While the system inherently avoids the major pitfalls of DSB-based editing, rigorous profiling using a combination of WGS, targeted sequencing, and in silico prediction is the standard for validating its specificity. The advent of machine learning tools like PRIDICT2.0 provides powerful support for the design phase, while benchmarked high-efficiency platforms enable more sensitive phenotypic screening [17] [31]. The most significant recent breakthrough is the engineering of next-generation editors (pPE, vPE) that proactively address the mechanistic source of on-target indels, resulting in dramatically improved edit-to-indel ratios [62].
Future directions in specificity analysis will likely involve the development of even more sensitive and scalable in vivo off-target detection methods. Furthermore, as single-prime-editing systems designed to treat multiple genetic diseases—such as the PERT system for nonsense mutations—move toward the clinic, the requirement for comprehensive, therapeutic-grade off-target profiles will become even more critical [12]. The continuous refinement of both the editing tools and the analytical frameworks for assessing them ensures that prime editing will remain at the forefront of precise genetic engineering for research and therapy.
Prime editing represents a transformative advancement in the field of genome engineering, offering unprecedented precision for modifying DNA without inducing double-strand breaks. This whitepaper delineates the current commercial landscape and key players driving the translation of prime editing from basic research to therapeutic applications. The market for genome editing, valued at $10.8 billion in 2025 and projected to reach $23.7 billion by 2030, is experiencing rapid growth fueled by technological innovation and increasing investment [63]. Within this expansive market, prime editing is emerging as a leading precision technology, with a specific market segment expected to grow at a notable CAGR of 24.1% from 2024 to 2031 [64]. The industry is concentrated in North America, particularly in U.S. biotech hubs like Massachusetts and California, which collectively host over 60% of the 332 identified gene-editing companies [65]. Major players such as Prime Medicine, Beam Therapeutics, and nChroma Bio are pioneering therapeutic development, while recent milestones, including the first FDA-approved clinical trial for a prime editing therapy in 2024, underscore the technology's immense clinical potential [64] [66]. This analysis provides researchers and drug development professionals with a comprehensive overview of the commercial ecosystem, key experimental methodologies, and essential research tools propelling the prime editing field forward.
Technical Foundations of Prime Editing
Prime editing is a versatile "search-and-replace" genome editing technology that directly writes new genetic information into a target DNA site without causing double-strand breaks (DSBs) [1]. This foundational characteristic addresses a primary limitation of earlier CRISPR-Cas9 systems, which rely on DSBs and error-prone cellular repair processes that can lead to unintended insertions, deletions, or chromosomal rearrangements [66]. The precision of prime editing makes it particularly suitable for therapeutic applications where minimizing off-target effects is critical.
The core prime editing machinery consists of three key components:
- A prime editor protein: This is a fusion protein comprising a Cas9 nickase (H840A) that cuts only a single DNA strand, fused to an engineered reverse transcriptase (RT) enzyme [1].
- A prime editing guide RNA (pegRNA): This specialized guide RNA performs two functions. It contains a spacer sequence that directs the complex to the target genomic locus, and it carries an extended segment that serves as a reverse transcription template (RTT), encoding the desired genetic edit [1].
The mechanism of prime editing follows a coordinated multi-step process [1]:
- The pegRNA guides the prime editor complex to the target DNA sequence.
- The Cas9 nickase nicks the non-target DNA strand, exposing a 3'-hydroxyl group.
- This exposed end serves as a primer for the reverse transcriptase, which uses the RTT on the pegRNA to synthesize a DNA flap containing the desired edit.
- Cellular repair machinery resolves this intermediate structure, favoring the incorporation of the edited strand into the genome, thereby permanently installing the new genetic sequence.
Evolution of Prime Editing Systems
Since its initial development, prime editing has undergone significant enhancements to improve its efficiency and versatility. The following table summarizes the evolution of prime editor systems, each building upon the last to achieve higher editing frequencies.
Table 1: Evolution of Prime Editing Systems
| Editor Version | Key Components | Editing Frequency (in HEK293T cells) | Major Innovation |
|---|---|---|---|
| PE1 [1] | Nickase Cas9 (H840A), M-MLV RT, pegRNA | ~10-20% | Proof-of-concept for "search-and-replace" editing. |
| PE2 [1] | Nickase Cas9 (H840A), engineered M-MLV RT, pegRNA | ~20-40% | Optimized reverse transcriptase for greater efficiency and stability. |
| PE3 [1] | PE2 system + additional sgRNA to nick non-edited strand | ~30-50% | Dual nicking strategy to encourage use of the edited strand as a repair template. |
| PE4 [1] | PE2 system + dominant-negative MLH1 (MLH1dn) | ~50-70% | Inhibition of the mismatch repair (MMR) pathway to boost editing efficiency. |
| PE5 [1] | PE3 system + dominant-negative MLH1 (MLH1dn) | ~60-80% | Combines dual nicking with MMR inhibition for maximal efficiency. |
| PE6 [1] | Compact RT variants, enhanced Cas9 variants, epegRNAs | ~70-90% | Improved delivery and pegRNA stability with engineered pegRNAs (epegRNAs). |
Diagram 1: The evolution of prime editing systems, highlighting key innovations that increased editing efficiency. PE4/5 introduced MMR inhibition (green), and PE6 introduced compact systems for delivery (red).
Commercial Landscape and Key Players
The gene editing industry is experiencing robust growth, with the global market projected to expand from $10.8 billion in 2025 to $23.7 billion by 2030, reflecting a compound annual growth rate (CAGR) of 16.9% [63]. The specific segment encompassing prime editing and CRISPR is growing even faster, with an estimated CAGR of 24.1% from 2024 to 2031 [64]. This growth is driven by rising demand for precision medicine, technological advancements, and increasing investment from both public and private sectors.
Global Distribution of Companies
The industry is globally distributed but concentrated in key innovation hubs. A 2025 analysis identified 332 gene-editing companies across 23 countries, with the United States dominating the landscape [65].
Table 2: Global Distribution of Gene Editing Companies (as of February 2025)
| Country/Region | Number of Companies | Key Characteristics and Hubs |
|---|---|---|
| United States | 217 | Dominated by hubs in Massachusetts (63 companies) and California (68 companies) [65]. |
| China | 30 | A rapidly growing hub focused on therapeutic applications and CRISPR technologies [65]. |
| United Kingdom | 16 | Strong presence in genome editing tools and therapeutics [65]. |
| Germany | 13 | Focused on gene editing for regenerative medicine and cell therapies [65]. |
| Rest of Europe | 25 | Includes France (8), Netherlands (7), Switzerland (5), and Ireland (5) with a growing presence [65]. |
Key Players and Strategic Focus
The commercial ecosystem can be segmented into companies developing therapeutics, providing tools and services, and enabling manufacturing. The following table profiles leading companies with a significant focus on prime editing and other next-generation technologies.
Table 3: Key Companies in the Prime Editing and Advanced Gene Editing Landscape
| Company | Primary Focus | Key Technology / Approach | Recent Developments (2024-2025) |
|---|---|---|---|
| Prime Medicine [64] [66] | Therapeutics | Prime Editing | Co-founded by David Liu. FDA cleared first clinical trial for prime editing therapy for chronic granulomatous disease (CGD) in April 2024. Reported positive early data from this trial in 2025 [64] [66]. |
| Beam Therapeutics [67] [66] | Therapeutics | Base Editing | A leader in base editing, a complementary precision technology. Advancing programs for sickle cell disease (BEAM-101) and alpha-1 antitrypsin deficiency (BEAM-302). Raised $500 million in direct financing in 2025 [67]. |
| nChroma Bio (formed from merger of Chroma Medicine & Nvelops) [67] | Therapeutics | Epigenetic Editing & Delivery | Merged in 2024 to combine Chroma's epigenetic editing platform (turning genes on/off without altering DNA) with Nvelop's non-viral delivery technology. Announced $75 million in new funding and a lead program for hepatitis B/D [67]. |
| Intellia Therapeutics [67] [14] | Therapeutics | In vivo CRISPR/Cas9 | A leader in in vivo gene editing. Its LNP-delivered therapy for hereditary transthyretin amyloidosis (hATTR) has shown ~90% protein reduction in Phase I. Advancing a Phase III trial for hereditary angioedema (HAE) [14]. |
| Caribou Biosciences [67] | Therapeutics | CRISPR-based Allogeneic Cell Therapies | Utilizes a chRDNA (CRISPR hybrid RNA-DNA) platform for allogeneic CAR-T therapies (e.g., CB-010, CB-011). In 2025, the company prioritized its pipeline and reduced its workforce by 32% to focus resources [67]. |
| Integrated DNA Technologies (IDT) [64] | Research Tools & Reagents | CRISPR and Editing Reagents | A key vendor providing essential reagents for gene editing research. In late 2025, launched the Alt-R HDR Enhancer Protein to improve knock-in efficiency in CRISPR experiments [64]. |
Prime Editing in Action: PERT as a Case Study
A landmark study from David Liu's lab at the Broad Institute exemplifies the innovative application of prime editing. The researchers developed a prime editing-mediated readthrough of premature termination codons (PERT) system, a disease-agnostic strategy to treat a wide class of nonsense mutations [12]. Nonsense mutations introduce a premature stop signal in a gene, causing the cell to produce a truncated, non-functional protein. They are responsible for approximately 30% of all rare diseases [12].
PERT Experimental Workflow and Methodology
The PERT strategy creatively uses prime editing not to correct the diverse nonsense mutations directly, but to install a universal "suppressor" tRNA gene into the cell's genome. This engineered tRNA enables the cellular machinery to read through premature stop codons, allowing for the production of full-length, functional proteins [12].
The detailed experimental protocol is outlined below:
Diagram 2: The PERT (Prime Editing-mediated Readthrough) experimental workflow, from tRNA engineering to functional protein production.
Key Experimental Models and Validation: The PERT system was rigorously validated in both in vitro and in vivo models [12]:
- Cell Models: The research team used human cell models of Batten disease, Tay-Sachs disease, and Niemann-Pick disease type C1. After PERT treatment, they observed restoration of enzyme activity to 20-70% of normal levels, a range considered potentially therapeutic [12].
- Animal Model: The system was tested in a mouse model of Hurler syndrome. Analysis of brain, liver, and spleen tissue showed that PERT restored approximately 6% of normal enzyme activity, which was sufficient to nearly eliminate all disease signs in the model [12].
The study reported no detected off-target edits or significant disruption of normal protein synthesis, underscoring the precision and safety of the approach [12].
The Scientist's Toolkit: Essential Research Reagents
Translating prime editing from concept to clinic relies on a suite of specialized research reagents and tools. The following table details key materials essential for conducting prime editing experiments.
Table 4: Essential Research Reagents for Prime Editing
| Research Reagent / Tool | Function in Prime Editing | Specific Examples / Notes |
|---|---|---|
| Prime Editor Plasmids | Serve as the DNA vector for expressing the prime editor fusion protein (nCas9-Reverse Transcriptase) in cells. | Plasmids for PE2, PE5, and PE6 systems are common starting points for research, each offering different levels of baseline efficiency [1]. |
| pegRNA Synthesis Tools | Critical for designing and producing the pegRNA, which specifies both the target site and the desired edit. | Services and software from vendors like Integrated DNA Technologies (IDT) and GenScript are widely used. Enhanced pegRNAs (epegRNAs) with structural motifs to reduce degradation are now standard for improved performance [1] [64]. |
| Delivery Vehicles | Facilitate the transport of prime editing components (mRNA, protein, or plasmid) into target cells. | Lipid Nanoparticles (LNPs) are highly effective for in vivo delivery to the liver [14]. Viral vectors (AAV, lentivirus) are also common. The choice of vehicle is a major determinant of editing efficacy [66]. |
| HDR Enhancers | Chemical or protein-based reagents that can improve the efficiency of homology-directed repair pathways, which can benefit prime editing outcomes. | IDT's Alt-R HDR Enhancer Protein is a commercially available solution designed to boost knock-in and editing efficiency in difficult-to-edit cells [64]. |
| Next-Generation Sequencing (NGS) Assays | Essential for quantifying on-target editing efficiency and comprehensively screening for potential off-target effects. | Whole-genome sequencing and targeted deep sequencing are mandatory for pre-clinical validation of editing precision and safety [12] [1]. |
Prime editing has firmly transitioned from a groundbreaking academic discovery to a validated technology with a rapidly maturing commercial ecosystem. The landscape is characterized by robust market growth, significant concentration of expertise in North American biotech hubs, and a dynamic mix of companies pursuing therapeutic, tool, and platform development. The progression of prime editing systems from PE1 to PE6 demonstrates a relentless focus on enhancing efficiency and specificity, while innovative strategies like the PERT system highlight the potential for a single editing agent to address multiple genetic diseases.
For researchers and drug development professionals, the path forward is clear yet challenging. Success in this field will depend on continued optimization of delivery systems, further refinement of editor efficiency and specificity, and navigating the evolving regulatory pathways. As clinical milestones accumulate and investment continues, prime editing is poised to play a central role in the next generation of precision genetic medicines, offering hope for curative treatments for a vast array of genetic disorders.
Conclusion
Prime editing represents a paradigm shift in genome engineering, offering unprecedented precision and versatility by avoiding double-strand breaks and enabling a wide range of edits. The technology has rapidly evolved through multiple generations, demonstrating therapeutic proof-of-concept in relevant disease models and is now entering clinical trials. While challenges in efficiency and delivery persist, ongoing engineering efforts continue to optimize the system. Its potential is further amplified by innovative, disease-agnostic strategies like PERT, which could allow a single therapy to treat multiple genetic disorders. For researchers and drug developers, prime editing is a powerful tool for functional genomics and a promising platform for developing a new class of one-time curative genetic therapies, poised to address thousands of genetic diseases with high precision.
References
- 1. Emerging trends in prime editing for precision genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322275/]
- 2. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 3. Prime Editing as a Precision Gene Editing Tool [https://www.synthego.com/guide/crispr-methods/prime-editing/]
- 4. Explainer: What Is Prime Editing and What Is It Used For? [https://crisprmedicinenews.com/news/explainer-what-is-prime-editing-and-what-is-it-used-for/]
- 5. Prime editing: therapeutic advances and mechanistic insights [https://pmc.ncbi.nlm.nih.gov/articles/PMC11946880/]
- 6. A benchmarked, high-efficiency prime editing platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10996517/]
- 7. Prime Editing Brings Precision and Breadth to Genome Editing [https://www.the-scientist.com/prime-editing-brings-precision-and-breadth-to-genome-editing-73253]
- 8. Prediction of prime editing insertion efficiencies using ... [https://www.nature.com/articles/s41587-023-01678-y]
- 9. Emerging trends in prime editing for precision genome ... [https://www.nature.com/articles/s12276-025-01463-8]
- 10. Optimized Ribonucleoprotein Complexes Enhance Prime ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12345536/]
- 11. A more precise way to edit the genome [https://news.mit.edu/2025/more-precise-way-edit-genome-0917]
- 12. Single prime editing system could potentially treat multiple ... [https://www.broadinstitute.org/news/single-prime-editing-system-could-potentially-treat-multiple-genetic-diseases]
- 13. Prime editing-installed suppressor tRNAs for disease ... [https://www.nature.com/articles/s41586-025-09732-2]
- 14. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 15. Designing and executing prime editing experiments in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9799714/]
- 16. Current advancement in the application of prime editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC9978821/]
- 17. A benchmarked, high-efficiency prime editing platform for ... [https://www.nature.com/articles/s41592-024-02502-4]
- 18. Prime Editing for Crop Improvement: A Systematic Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12385346/]
- 19. Prime Editing [https://www.bioconductor.org/packages//release/bioc/vignettes/multicrispr/inst/doc/prime_editing.html]
- 20. Prime Editing Technology and Its Prospects for Future ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10530660/]
- 21. Protocol for the design, conduct, and evaluation of prime ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10519846/]
- 22. Frontiers | Systematic optimization of prime for enhanced... editing [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1589034/full]
- 23. Systematic optimization of prime editing for enhanced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12069386/]
- 24. Single Prime Editing Platform Shows Potential vs. Multiple ... [https://www.genengnews.com/topics/genome-editing/single-prime-editing-platform-shows-potential-vs-multiple-diseases/]
- 25. : An All-Rounder for Genome Prime - PMC Editing Editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC9456398/]
- 26. : Adding Precision and Flexibility to CRISPR Prime Editing Editing [https://blog.addgene.org/prime-editing-crisp-cas-reverse-transcriptase]
- 27. - Wikipedia Prime editing [https://en.wikipedia.org/wiki/Prime_editing]
- 28. PrimeDesign software for rapid and simplified design of prime ... editing [https://www.nature.com/articles/s41467-021-21337-7?error=cookies_not_supported&code=f57306c1-fc26-42f7-bd59-7c1f085f93ee]
- 29. TIGER: A tdTomato in vivo genome-editing reporter mouse for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12415246/]
- 30. In vivo precision base editing to rescue mouse models of ... [https://www.sciencedirect.com/science/article/pii/S2162253125001763]
- 31. Machine learning prediction of prime editing efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7617539/]
- 32. Single prime editing system could potentially treat multiple ... [https://www.einpresswire.com/article/868636956/single-prime-editing-system-could-potentially-treat-multiple-genetic-diseases]
- 33. How to fix genetic 'nonsense': versatile gene-editing tool ... [https://www.nature.com/articles/d41586-025-03770-6]
- 34. Advances and optimization strategies in prime editing of ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925002562]
- 35. Prime Editing: The Next Frontier in Precision Gene Therapy [https://pubmed.ncbi.nlm.nih.gov/41038616/]
- 36. Search-and-replace genome editing without double-strand ... [https://www.nature.com/articles/s41586-019-1711-4]
- 37. World first: ultra-powerful CRISPR treatment trialled in a ... [https://www.nature.com/articles/d41586-025-01593-z]
- 38. Prime Medicine Announces Breakthrough Clinical Data ... [https://investors.primemedicine.com/news-releases/news-release-details/prime-medicine-announces-breakthrough-clinical-data-showing]
- 39. Design of prime-editing guide RNAs with deep transfer learning | Nature Machine Intelligence [https://www.nature.com/articles/s42256-023-00739-w]
- 40. Systematic pegRNA design with PRIDICT2.0 and ePRIDICT for efficient prime editing | Nature Protocols [https://www.nature.com/articles/s41596-025-01244-7]
- 41. Mismatch prime editing gRNA increased efficiency and reduced indels | Nature Communications [https://www.nature.com/articles/s41467-024-55578-z]
- 42. EXPERT expands prime editing efficiency and range of ... [https://www.nature.com/articles/s41467-025-56734-9]
- 43. Rapid generation of long, chemically modified pegRNAs for prime editing | Nature Biotechnology [https://www.nature.com/articles/s41587-024-02394-x]
- 44. Enhanced prime editing systems by manipulating cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8584034/]
- 45. Improving prime editing by studying DNA repair [https://frontlinegenomics.com/improving-prime-editing-by-studying-dna-repair/]
- 46. Prime Editing and DNA Repair System [https://www.mdpi.com/2073-4409/13/10/858]
- 47. Exonuclease-enhanced prime editors [https://www.nature.com/articles/s41592-023-02162-w]
- 48. Prime editing: therapeutic advances and mechanistic insights [https://www.nature.com/articles/s41434-024-00499-1]
- 49. Design Tips for Prime Editing [https://blog.addgene.org/design-tips-for-prime-editing]
- 50. Comparative Analysis of Methods for Assessing On-Target ... [https://www.mdpi.com/2409-9279/8/2/23]
- 51. Harnessing artificial intelligence to advance CRISPR ... [https://www.nature.com/articles/s41576-025-00907-1]
- 52. Comprehensive analysis of prime editing outcomes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8789035/]
- 53. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 54. Prime Editing: Mechanistic Insights and DNA Repair ... [https://www.mdpi.com/2073-4409/14/4/277]
- 55. Prime editing: Mechanism insight and recent applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10754014/]
- 56. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 57. CRISPR Base Editing Explained | ABE & CBE in Gene ... [https://www.synthego.com/crispr-base-editing-guide/]
- 58. Base editing: a brief review and a practical example - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8038524/]
- 59. Explainer: What Are Base Editors and How Do They Work? [https://crisprmedicinenews.com/news/explainer-what-are-base-editors-and-how-do-they-work/]
- 60. Increasing the Targeting Scope of CRISPR Base Editing ... [https://pubmed.ncbi.nlm.nih.gov/35238621/]
- 61. ProPE expands the prime editing window and enhances ... [https://www.nature.com/articles/s41929-025-01406-6]
- 62. Engineered prime editors with minimal genomic errors [https://www.nature.com/articles/s41586-025-09537-3]
- 63. Genome Editing Market Report 2025: CRISPR and Beyond [https://finance.yahoo.com/news/genome-editing-market-report-2025-151500990.html]
- 64. Prime Editing and CRISPR Market to Grow at 24.1% CAGR ... [https://www.einpresswire.com/article/866862036/prime-editing-and-crispr-market-to-grow-at-24-1-cagr-through-2031-datam-intelligence]
- 65. Gene Editing Companies Landscape in 2025: Who's Worth ... [https://www.samps.org/blog/gene-editing-landscape-2025]
- 66. David Liu Wins 2025 Breakthrough Prize for Base Editing and ... [https://www.the-scientist.com/david-liu-wins-2025-breakthrough-prize-for-base-editing-and-prime-editing-72888]
- 67. Ten CRISPR companies to watch in 2025 [https://www.labiotech.eu/best-biotech/crispr-companies/]